
在R中使用LIME解释机器学习模型
作者:PURVAHUILGOL
翻译:陈丹
校对:欧阳锦
关键词:机器学习模型解释、R语言、LIME
概述
仅仅构建模型但无法解释它的输出结果是不够的。
本文中,要明白如何在R中使用LIME来解释你的模型。
介绍
我曾经认为花几个小时来预处理数据是数据科学中最有价值的事情。这是我还作为一个初学者时的误解。现在,我意识到,能向一个对机器学习或其他领域的行话不太了解的外行解释你的预测和模型才更有意义。
考虑一下这个场景——你的问题陈述涉及预测患者是否患有癌症。千辛万苦,你获得和清理了数据、为它构建了模型,并经过大量的努力、实验和超参数调整,你达到了90%以上的精度。太棒了!你走到医生跟前告诉他,你可以90%肯定地预测病人是否得了癌症。
然而,医生问的一个问题会把你难倒:“每个病人都不同,并且会有很多参数可以决定肿瘤的良恶性,我和病人要怎么相信你的预测?”
这就是模型可解释性问题的由来——如今,有多种工具可以帮助你高效地解释模型和模型预测,而不必深入探究模型的各种细节。这些工具包括SHAP、Eli5、LIME等。今天,我们将讨论LIME。
在本文中,我将解释LIME以及在R中它如何使解释模型变得容易。
什么是LIME?
LIME全称是“Local InterpretableModel-Agnostic Explanations”(局部可解释的模型无关阐释)。2016年首次提出LIME技术的论文被它的作者,MarcoTulio Ribeiro、SameerSingh和CarlosGuestrin,恰当地命名为《“WhyShould I Trust You?” Explaining the Predictions of Any Classifier》(《“为什么我应该相信你?”解释任何分类器的预测》)。

基于这一基本但至关重要的信任原则,LIME背后的理念是回答每个预测和整个模型是“为什么”。LIME的创造者列出了四个必须满足的基本准则:
可解释性:对预测的解释应该是可以理解的,即可由目标人群解释。
局部准确性:我们应该能够解释个体的预测。作者称之为“local fidelity”。
模型无关性:解释方法应适用于所有模型。这被作者称为解释是“model-agnostic”。
整体解释性:除了个体预测外,模型还应具有整体解释性,即应考虑全局视角。
LIME如何工作?
进一步展开LIME的工作原理发现,其背后的主要假设是,每个模型在局部尺度上都像一个简单的线性模型,即在单个行级别的数据上。即使这篇论文和作者并不打算证明这一点,但我们可以感知,在个体水平上,我们可以在行上拟合这个简单模型,它的预测将非常接近我们复杂模型对该行的预测。很有趣,不是吗?
此外,LIME还扩展了这一现象,即围绕这一行中的小变化来拟合这些简单模型,然后通过比较简单模型和复杂模型对该行的预测来提取重要特征。
LIME既适用于表格/结构化数据,也适用于文本数据。
你可以在这里阅读更多关于LIME如何使用Python的内容
(https://www.analyticsvidhya.com/blog/2017/06/building-trust-in-machine-learning-models/),本文中我们将介绍如何使用R。
所以启动你的Notebooks或Rstudio,让我们开始吧!
在R中使用LIME
第一步:安装LIME和其他所有这个项目所需要的包。如果你已经安装了它们,你可以跳过这步,从第二步开始。
install.packages('lime')install.packages('MASS')install.packages("randomForest")install.packages('caret')install.packages('e1071')
第二步:安装好这些包后,我们先导入它们:
library(lime)library(MASS)library(randomForest)library(caret)library(e1071)
由于我们用解释病人是否患有癌症的预测作为例子,我们将使用活检数据集。这个数据集包含了699名患者及其乳腺癌肿瘤活检的信息。
第三步:我们导入这些数据并看看前几行数据:
data(biopsy)
第四步:数据勘探
4.1 由于ID列只是一个标识符,并没有用,因此我们首先将它移除:
biopsy$ID<- NULL4.2 让我们重新命名剩下的列,这样当我们使用LIME来理解预测结果的可视化解释过程中,我们能够清晰了解特征的名称:
names(biopsy) <- c('clumpthickness', 'uniformity cell size', 'uniformity cell shape', 'marginaladhesion', 'single epithelial cell size', 'bare nuclei', 'bland chromatin','normal nucleoli', 'mitoses','class')4.3 接下来,检查是否有缺失值。如果有,在进一步处理前,我们应先处理它们:
sum(is.na(biopsy))4.4 我们现在有两种选择:要么既可以补全这些值,要么也可以使用na.omit函数直接丢掉包含缺失值的行。由于本文不涉及清理数据的内容,因此我们将使用后一种方法。
biopsy <-na.omit(biopsy)sum(is.na(biopsy))
最后,让我们看看前几行数据来确认我们的数据表。
head(biopsy,5)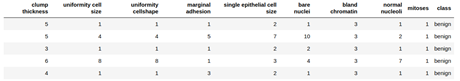
第五步:将这些数据分为训练集和测试集,并检查数据的维度。
# 75% of thesample sizesmp_size <-floor(0.75 * nrow(biopsy))## set theseed to make your partition reproducible - similar to random state in Pythonset.seed(123)train_ind<- sample(seq_len(nrow(biopsy)), size = smp_size)train_biopsy<- biopsy[train_ind, ]test_biopsy<- biopsy[-train_ind, ]
检查维度:
cat(dim(train_biopsy),dim(test_biopsy))因此,在训练集中有512行,测试集中有171行数据。
第六步:我们将通过caret包使用随机森林模型。我们也不会调试超参数,只是实现一个5次10折的交叉验证和一个基础的随机森林模型。所以在我们训练集上训练和拟合模型时,不要进行干预。
我鼓励你们也可以用这些参数来试验其他模型。
model_rf <- caret::train(class~ ., data = train_biopsy,method = "rf", #random foresttrControl = trainControl(method ="repeatedcv", number = 10,repeats = 5, verboseIter = FALSE))
让我们看看模型总结:
model_rf
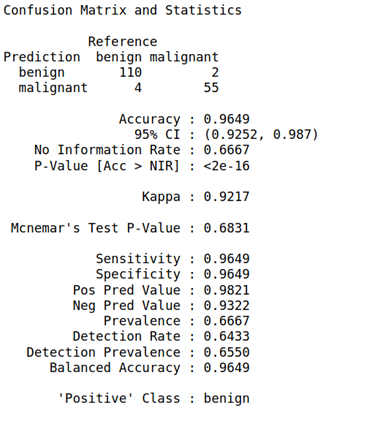
第七步:我们将把预测函数运用到我们的测试集上,并且建立一个混淆矩阵:
biopsy_rf_pred<- predict(model_rf, test_biopsy)confusionMatrix(biopsy_rf_pred,as.factor(test_biopsy$class))
第八步:有了模型后,我们将用LIME来创造一个“explainer”实体。这个实体也与我们将使用来查看解释的其他LIME函数有关。
就像训练模型并拟合数据一样,我们也使用lime() 函数来训练explainer,然后使用explainer()来得到新的预测结果。
explainer<- lime(train_biopsy, model_rf)让我们只使用5个特征来阐释从测试集得到的5个观测值。可以随意测试任何多个特征参数。这一步也可以跳过。
整个测试集,或测试集的单一行
explanation <-explain(test_biopsy[15:20, ], explainer, n_labels = 1, n_features = 5)其他你可以试验的参数有:
1. n_permutation: 用于每次解释的置换次数。
2. feature_select: 用于挑选特征的算法。我们可以从以下6个中选择:
auto:如果n_features小于等于6,则使用“forward_selection”;,否则,使用“highest_weights”。
none:忽略n_features并且使用所有特征。
forward_selection:根据脊回归模型的质量,每次添加一个特征直到达到n_features。
highest_weights:拟合一个脊回归模型并选择一个n_features作为最高绝对权重。
lasso_path:拟合一个lasso模型并选择一个最小角回归路径最后收敛到0的n_features。
tree:拟合一个树来选择n_features(需要是2的次方)。需要XGBoost的最新版本。
3. dist_fun:距离函数。我们将使用这个函数来比较我们的局部模型对某一行的预测和全局模型(随机森林)对该行的预测。默认是高尔距离,但我们也可以使用欧氏距离、曼哈顿距离等等。
4. kernel_width:个体置换预测和全局预测的距离如上计算,并转换成一个相似度评分。
第九步:让我们来可视化这些解释以更好地理解它们:
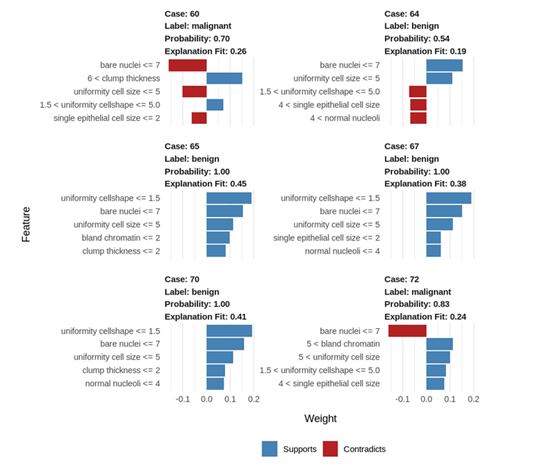
如何解释这些结果?
1. 蓝/红色:正相关的特征标记为蓝色,负相关的特征标记为红色。
2. Uniformity cell shape(细胞形状的均匀性)<=1.5:更低的值与良性肿瘤正相关。(值越低,良性肿瘤概率更高)
3. Bare nuclei(裸核)<= 7:更低的值与恶性肿瘤负相关。(值越低,恶性肿瘤概率更低)
4. 65、67和70号案例很相似,但是64号良性案例有不一样的参数。
5. 在这种情况下,细胞形状的均匀性和单个上皮细胞的大小是异常的。
6. 尽管有这些偏离值,这个肿瘤依旧是良性的,这表明这个病例的其他参数值补偿了这种异常。
让我们也来可视化一个案例的所有特征:
explanation <-explain(test_biopsy[93, ], explainer, n_labels = 1, n_features = 10)plot_features(explanation)
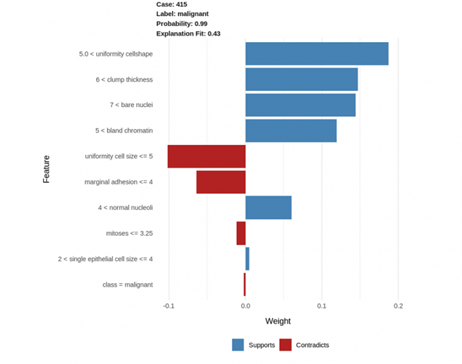
Uniformity cellshape(细胞形状的均匀性) > 5.0:高的值与恶性肿瘤正相关(这个值越高,肿瘤恶性的可能性更大)。
Clump thickness(团块厚度) > 6.0:高团块厚度值与恶性肿瘤正相关。
同样的,bare nuclei(裸核) > 7.0 以及 bland chromatin(布兰德染色质)> 5.0 与恶性肿瘤正相关。
相反,uniformity of cell size(细胞大小的均一性)<= 5.0 和marginaladhesion(边缘附着力)<= 4:这2个参数低值与恶性肿瘤的恶性成正相关与恶性肿瘤负相关[1] 。因此,这些值越低,肿瘤恶性的可能性越小。
因此,从上可知,我们可以得出结论,这些参数高值能够表明肿瘤有更高可能性是恶性。
我们也可以通过看这行真实的数据来确定上述的解释:

尾注
最后,我们探讨了LIME以及如何使用它来解释我们模型的个体结果。这些解释更好地帮助我们传达自己的想法有助于更好地讲故事,并帮助我们向一个可能拥有领域专业知识,但没有模型构建技术诀窍的人解释为什么模型会做出某些预测。而且,使用它非常容易,在我们有了最终的模型之后只需要几行代码。
然而,这并不是说LIME没有缺点。我们使用的LIME-Cran包并不是我们在本文中介绍的原始Python实现的直接复刻,因此,它不像Python那样支持图像数据。另一个缺点是,局部模型可能并不总是准确的。
我期待着使用不同数据集和模型来更多地探索LIME,并且探索R中的其他技术。你在R中使用了哪些工具来解释你的模型?一定要在下面分享你如何使用他们以及你使用LIME的经历!
这里应该也是正相关吧。值越低,越不容易产生恶性肿瘤。
原文标题:
PolynomialRegression — Gradient Descent from ScratchMLInterpretability using LIME in R
原文链接:
https://www.analyticsvidhya.com/blog/2021/01/ml-interpretability-using-lime-in-r/
推荐阅读:
译者简介

陈丹,复旦大学大三在读,主修预防医学,辅修数据科学。对数据分析充满兴趣,但初入这一领域,还有很多很多需要努力进步的空间。希望今后能在翻译组进行相关工作的过程中拓展文献阅读量,学习到更多的前沿知识,同时认识更多有共同志趣的小伙伴!