
空间转录组学
空间转录组学
《Nature Methods》杂志将空间转录组学评选为2020年Method of the Year!
0
Nature Methods

星系探索。在转录组学领域,空间转录组也开辟了新世界。Credit: bjdlzx/Getty Images
Nature Methods在今年1月份将空间转录组学评价为2020年度方法,空间转录组方法最近今年越来越受到欢迎,它将带领组学在生物学的研究上进入新的阶段。从转录组测序到实现单细胞转录测序,这一过程为精确认识不同细胞类型提供了巨大的帮助。现在,新的空间转录组方法不仅带来转录组信息,同时还提供空间信息,帮研究者更好辨别转录的位置,进一步扩展到单细胞分辨率,这会更大程度提升科学家对单个细胞的认识和解读。这个过程中涌现了一大批新的技术方法,新的成像技术以及把相关科研成果转化的生物技术公司,面临的挑战也是巨大的,尤其是如何处理产生的数据,如何更好的提升现有方法让更多的人在自己的实验室去实现,如何把这些技术商业化等。作者通过与不同的人的交流访谈,对上述问题进行了深切解读。
首先我们需要为大量 RNA 测序准备样本,其中组织被均质化并分析以产生组织细胞中 mRNA 的平均基因表达——称为转录组。之后开发单细胞 RNA 测序 (scRNA-seq) ,对每个细胞的转录组进行更精细的评估。在 scRNA-seq 中,研究人员将细胞从组织中脱离出来,从而可以根据基因表达来辨别细胞类型。艾伦脑科学研究所(Allen Institute for Brain Science)所长Hongkui Zeng说,处理单个细胞更像是在研究水果沙拉(fruit salad)而不是冰沙(smoothie)。现在,通过空间解析转录组学方法,科学家可以获得转录组数据并了解这些细胞在组织中的空间位置。曾与Zeng共同接受采访的艾伦研究所研究员 Bosiljka Tasic 说:“水果馅饼(fruit tart)才是是空间转录组学(不是水果沙拉),你确切地知道每块水果的位置以及各水果之间的关系。”考虑到人们可以从单个细胞和空间分辨细胞中学到多少东西,“smoothie”方法(RNAseq)很快就过时了。空间分析提供所有细胞的转录组信息还不是常规做法,该领域正在朝着单细胞方向快速发展。卡罗林斯卡学院(Karolinska Institute)生物学家 Sten Linnarsson 说,“一切都与空间有关。空间单细胞转录组学是单细胞分析之后的下一波浪潮,对于研究人类疾病的实验室特别有用,人类疾病通常始于单细胞并在空间上传播。我想,几年后我们将在活体组织中进行空间单细胞组学实验,”这将单细胞发展带入一个完整的循环。
1
Welcome, family
热衷于在空间位置中量化 mRNA 的实验室采用了一系列方法,这些方法可在神经科学、癌症研究或发育生物学等领域提供空间解析的转录组数据。“我认为未来是光明的,”华盛顿大学(University of Washington)的 Jay Shendure 说,他最兴奋的是如何应用这些方法来获得结合细胞状态、历史和命运的空间解析发育图谱。这是一个非常活跃的领域,这种技术开辟了新的实验机会。哈佛大学(Harvard University)的Xiaowei Zhuang,同时也是霍华德休斯医学研究所(Howard Hughes Medical Institute)研究员,一直致力于影像学的研究。她和她的实验室开发了一种超分辨率显微镜方法,即随机光学重建显微镜 (STORM)[备注:随机光学重建显微法(stochastic optical reconstruction microscopy,STORM) 是一种将荧光光谱和显微分析技术应用于单个分子之上的崭新的物理手段,其是一种比传统光学显微镜高10倍以上分辨率的新型显微技术] 。
当单细胞基因组学出现时,Zhuang致力于将成像和基因组学结合起来,以“获得两全其美”。从物理学家转为生物学家的乌得勒支研究所(Hubrecht Institute)研究员Alexander van Oudenaarden 说,方法,尤其是从 2012 年左右开始出现的用于在单细胞序列捕获空间信息的方法,已经融合了以前独立的成像、测序和分子表征。他和他的实验室结合了单细胞和空间转录组学来比较原肠胚和小鼠胚胎中的基因表达。对单细胞的研究起源于 19 世纪,例如 Rudolf Virchow 观察到的疾病始于细胞,而不是整个生物体。十年前,当 van Oudenaarden 和其他人通过显微镜观察细胞在生长的生物体中的发育、生长和迁移时,“我们总是有点嫉妒测序的世界。”他和他的团队可以制作延时影相并捕捉复杂的过程,但只能用少量荧光蛋白标记基因表达。基因组学实验室可以测量数千个基因。为了在组织水平做到这一点,他们“将它们放入搅拌机中”。现在可以评估许多单个细胞和许多基因并跟踪细胞的组织环境、它们的空间属性。现在有各种聪明的方法来保存空间信息,这是一项非常令人兴奋的技术。
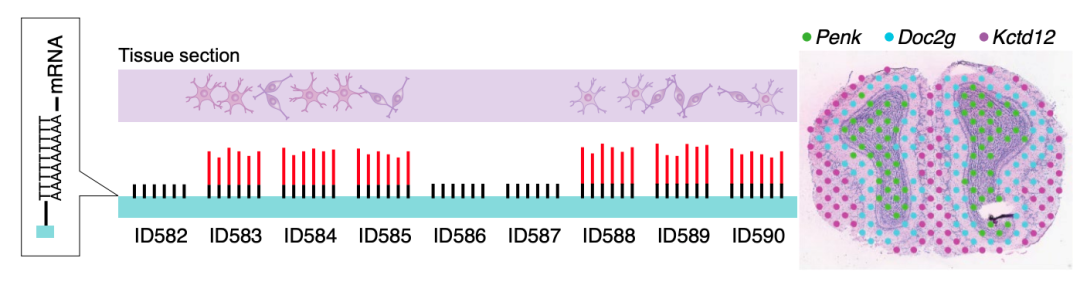
瑞典的研究人员开发了一种方法,对固定的染色组织进行成像、透化,然后将 mRNA 附着到一系列带条形码的寡核苷酸上。RNA 被逆转录cDNA,对cDNA 进行测序并产生空间解析的转录组信息。Credit: Adapted with permission from ref. 4, AAAS
正如来自 KTH 皇家理工学院生命科学实验室的 Joakim Lundeberg 及其同事所指出的,空间技术可以分为涉及对显微解剖组织进行基因表达分析的技术,以及涉及原位杂交、原位测序、原位捕获和空间数据的重构。Zeng说,单分子荧光原位杂交 (smFISH) 是带有空间技术的杂交方法的开端”。在这种方法中,多个寡核苷酸携带荧光标记并与 RNA 分子结合。来自艾伦研究所、生命科学实验室和卡罗林斯卡研究所的研究人员指出,smFISH检测灵敏度接近 100%。
来自 KTH 的 Patrik Ståhl 解释说,大约在 2009 年,卡罗林斯卡学院的研究员 Jonas Frisén 着手研究如何从典型的组织切片中获取更多信息,这些切片是用可追溯到 19 世纪的染色剂制备的切片。Frisén 联系了 KTH 的同事 Lundeberg,Ståhl 是他的博士生。Fredrik Salmén 是 KTH 的一名硕士生;Ståhl 后来加入 Frisén 实验室担任博士后研究员,Salmén 成为 Lundeberg 实验室的博士生。Ståhl 和 Salmén 的一个合作项目产生了一种空间分析方法,通过该方法对固定的染色组织进行成像,然后进行透化,释放的 mRNA 移动并附着在组织下方的阵列上,带有条形码的寡核苷酸,将它们固定在组织中的位置。逆转录后,组织被酶促反应去除,剩下的是连接到寡核苷酸阵列的空间条形码cDNA 分子,接下来是对这些 cDNA 的测序,其中位置条形码提供空间解析的转录组信息。科学家们想将此技术在单个细胞水平上实现。“我们真的很想达到那个水平,”Salmén 说,他与 Ståhl 共同接受了采访,他现在是 van Oudenaarden 实验室的博士后研究员。该方法的分辨率最终为 100 µm,即数十个细胞。Salmén 说,他们面临的众多技术挑战之一是 mRNA 可以向多个方向扩散,这可能会导致空间数据不准确或混合表达模式。他们开发了避免这种情况的方法。“这是一场漫长的斗争,”Ståhl 说,但他们的团队成功了,Ståhl 和 Salmén 共同成为论文的第一作者。Ståhl 说,这篇论文和过程塑造了他们的科学和他们的职业生涯。
Ståhl 说,Salmén 的关键想法是建立与荧光核苷酸的初始反应,使细胞的mRNA与阵列表面探针相遇的位置可见。Ståhl 说,这让您可以准确地想象捕获的 mRNA 合成 cDNA 的位置。“你会得到一个非常好的荧光足迹,显示所有东西的去向,对我们来说,这是让其他人开始工作的巨大基石,”他说。Salmén 笑着说,这个基石让许多人相信这种方法具有潜力——即使是“非信徒”。他表示,方法开发意味着很多失败。该方法通过Spatial Transcriptomics 公司商业化,该公司于 2018 年被 10x Genomics 收购,从而产生了产品 Visium。
空间技术通过定位表达的基因来帮助构建图谱。在这里,seqFISH+ 用于测量小鼠皮层中的 10,000 个基因。Credit: Cai lab, Caltech, I. Strazhnik; adapted with permission from ref. 6, Springer Nature.
2
Other spatial travels
加州理工学院(California Institute of Technology)研究员 Long Cai 说,成像本质上是空间的,而且是原生的 3D ,他的职业生涯也受到空间方法的影响。他说,要使用显微镜获得 3D 信息,“您只需进行切片”。他回忆起他的加州理工学院同事芭芭拉·沃尔德 (Barbara Wold) 向他提到 RNA 测序可以提供多么丰富的信息。那是十年前,第二代测序技术刚刚兴起。“仔细想想,测序实际上是一个盒子里的单分子成像”,他说,“测序仪可以产生 200 个碱基对的读数,这些读数来自 200 轮化学反应和成像”。基因组学方法产生细胞信息,但与 smFISH 相比,通过成像读取单细胞基因组信息需要更多的标签。Cai 应用超分辨率显微镜将可辨别的荧光点填充到成像细胞中。这些点可以是标记 RNA、DNA 或蛋白质。他实验室的方法 seqFISH5 和 seqFISH+ 使用一系列带有荧光团的 FISH 探针对 mRNA 进行连续轮次杂交,转录物固定在细胞中。在每一轮杂交中,探针都被去除,便于下一轮与不同荧光团的杂交,荧光信号序列提供空间解析的转录组数据。他说,“您可以直接原位完成,而不是将成像信息转换为测序读数。”他创立了一家名为 Spatial Genomics 的初创公司,旨在将 seqFISH 商业化。
在他看来,研究人员需要达到单细胞分辨率,scRNA-seq 提供了单细胞分辨率,但到目前为止,组织成像还没有。分辨率很重要,因为在一个感兴趣的区域,组织可能有多种细胞类型,但如果没有足够的转录组分辨率,就会出现“完全错误的图片”。如果一种方法效率低下,可能会遗漏一些信号,例如以 10 个或更少拷贝表达的转录因子的 mRNA。Cai认为当前许多空间方法的弱点是它们导致“最终你必须放入测序仪”。高通量基因组学已经有了一个良好的开端,对于测序现在可以做的几乎所有事情,都会有一种空间分析方法可以做同样的事情甚至做得更好。
博德研究所(Broad Institute)的医师兼科学家埃文·马科斯科 (Evan Macosko) 一直热衷于使用基因组学对细胞和组织进行新型测量。他在哈佛医学院 Steve McCarroll 实验室担任博士后时共同开发了Drop-seq[备注:一种scRNAseq建库方法]。使用 Drop-seq,准备数千个测序文库需要一天的时间。在微流体装置中,细胞穿过狭窄的通道,最终被包裹在一个带有珠子的液滴中,每个珠子都配备了一个独特的 12 碱基对寡核苷酸条形码。细胞在液滴中裂解,mRNA 被编码并逆转录成 cDNA,然后进行测序。Macosko 与时任 Broad 研究员的 Fei Chen 一起开发了 Slide-seq,后者共同发明了膨胀显微镜,现在在 Broad 拥有一个实验室。Macosko 说,该方法借鉴了“DNA 条形码策略和其他一些技巧”。随着 smFISH 的发展,该技术成为他实验室的主力军。多重原位杂交策略很有用,但需要时间和技术。“这不是传统分子生物学或细胞生物学实验室可以立即建立起来的,”他说,他寻求一种不同的方法,利用基因组学实验室中基于 Illumina 的测序的广泛基础设施,在全基因组范围内获得细胞或近细胞分辨率。他的团队应用了 Drop-seq 的条形码方案,从 50 微米的珠子开始,然后转向 10 微米的珠子。Macosko 说,Chen 实验室的 Sam Rodriques 和 Macosko 实验室的 Robert Stickels 想出了如何将珠子单层排列为二维阵列,并开发了一种将 RNA 转移到珠子的方案,这说起来容易做起来难。Slide-seq 的输出接近单细胞分析的输出,这使得使用单细胞计算工具成为可能。他们已经运行了单细胞分析工具在一些项目中,如 velocyto 和 Monocle,他们使用这种方法研究了晶状体和胚胎皮层的发育。较低分辨率的空间技术每个像素有太多的细胞,这使得去卷积更加困难。由于 Slide-seq 使用珠子,可以继续使珠子更小,分辨率更高。该团队一直致力于提高该方法的效率,Slide-seq v2 的 mRNA 捕获效率接近 scRNA-seq 技术。效率很重要,因为对于稀少的转录需要足够的信息来准确地将它们分配到特定位置。另外,关于将 Slide-seq 商业化的讨论还处于早期阶段。

庄小伟开发了超分辨率显微镜方法STORM。单细胞基因组学让她想到了将基因组学和成像融合起来。Credit: Harvard University
3
Compute that space
约翰霍普金斯大学(Johns Hopkins University)计算癌症生物学家 Elana Fertig 说,尽管在理想的世界中,她想要达到单细胞分辨率,但事实上,它不能完美地解析每个细胞。她致力于研究肿瘤异质性,空间技术可以帮助解决这些问题。她的一个项目研究 T 细胞浸润肿瘤,该项目专注于多组学,整合来自不同模式的数据,研究人员使用来自不止一种空间分析方法的数据。她发现 10x Genomics 的 Visium 技术比 FISH 更容易获得。分子变化可以定义肿瘤类型,但这些类别中的肿瘤可能差异很大。“细胞类型不是那么二元的【备注:这里她想表达细胞种类远比想象的复杂的多】,”她说,“她想知道迄今为止流式细胞术是否已经塑造了细胞分型。”对于这项工作,团队合作在开发数据分析和可视化方法方面变得越来越重要。Fertig 实验室是美国国家癌症研究所癌症研究信息技术计划的一部分,旨在构建用于解释越来越多地包含空间数据的多维组学数据的框架。随着每个细胞有更多的标记,数据的高维度增加了数据解释的挑战。“我认为这是空间领域目前面临的挑战,”她说。为了整合成像和单细胞数据进行空间转录分析,她和她的团队应用迁移学习来叠加基因相关特征并进行降维。
Macosko 预设了一个围绕寻找空间可变基因或寻找利用空间信息的轨迹计算开发方案。尽管付出了很多努力,但Shendure 表示,管理数据和跨平台集成仍然是一个巨大的挑战。每个实验都会产生大量数据,研究人员面临着关于保存什么和丢弃什么的决定,并且不想偏离基因组学中开放数据共享的原则。

空间解析转录组学揭示了在基于干细胞的发育模型中表达的体节发生标记,如图在小鼠中。Credit: A. van Oudenaarden, Hubrecht Inst.
4
Data juggling
在艾伦研究所,大脑中的细胞分型等大规模项目与处理大型数据集一样司空见惯。Tasic 说,这甚至让他们感到惊讶,当查看每个细胞具有数万个属性的单细胞和空间转录组数据时,他们能以多快的速度达到标准分析的界限。数据矩阵变得难以管理,他们与处理大规模数据矩阵的生物学之外的其他人合作。未来的计算之路既艰巨又令人兴奋。他在研究生院“测量所有单细胞中所有基因”的梦想开始实现。除了数据大小和维度之外,Zeng 说,他和他的团队致力于集成和校正批量效应。即使使用相同模式捕获的数据也可能会因批次而略有不同,因此必须进行标准化。为了整合成像、测序和电生理学等模式,他们构建了聚类方法并利用了不同类型的机器学习方法去尝试。
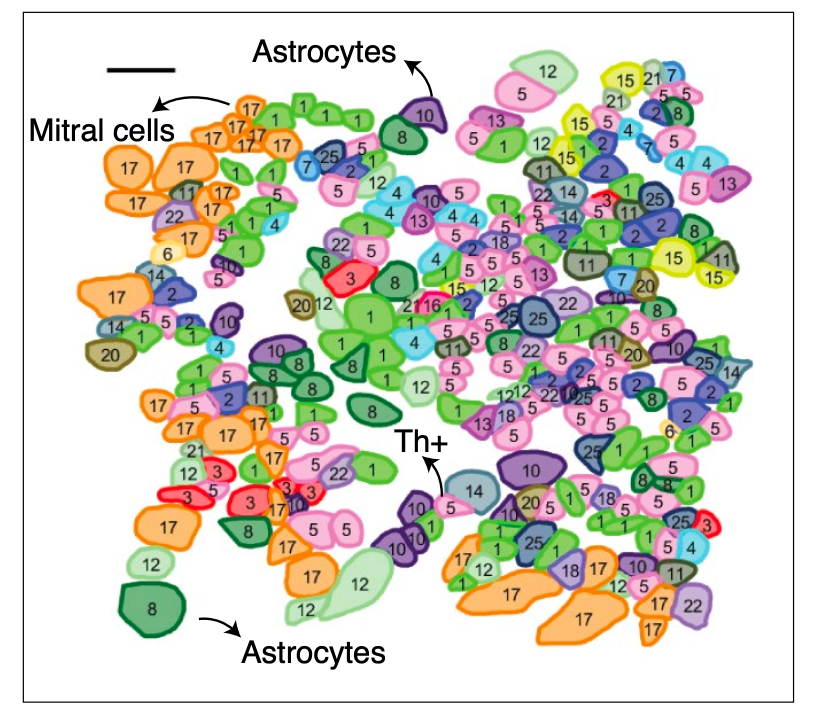
空间技术可以揭示组织的复杂细胞类型混合物——这里是小鼠大脑的嗅球。Credit: Cai lab, Caltech, I. Strazhnik Adapted with permission from ref. 6, Springer Nature.
van Oudenaarden 说,存在很多机会,可以通过计算将scRNA-seq 和空间信息结合起来,将单细胞映射回空间,并构建和使用参考图谱。他对从原位杂交或不使用以前图谱重建空间图谱的计算方法很感兴趣。一种这样的方法,novoSpaRc,在没有参考图谱的情况下进行“基因表达绘制图谱”。它基于关于基因表达如何在组织切片中变化的假设。van Oudenaarden 说,拥有更多关于组织的数据是一笔财富,这笔财富使得了解重要的“骨骼”(生物学的核心方面)成为“一个更大的难题”。对他来说,单细胞组学和空间方法的结合“确实是一种产生无限可能的方式,但还需要使用单独收集的数据来确认和验证发现。
5
Commercial takes
空间技术有着悠久的历史,Shendure 说,例如在 Mats Nilsson、George Church 和其他人的实验室中,他们已经在组织切片上直接进行基因分型或测序等项目十多年了。空间分析在过去一两年中蓬勃发展成为一个领域,部分是由于单细胞领域已经成熟,并且技术已转变为具有广泛用途的商业仪器。不过,还有一点仍然是挑战之一,许多最令人兴奋的方法仍然在某种程度上是定制的,比较局限,实际上其只能在一个或几个实验室中运行。当谈到迄今为止的空间方法时,他说,问题是如何让其他人更容易地采用这些方法,这些方法必须能够大规模推广。Zeng说,科学家所做的大部分工作既困难又昂贵,但技术发展有助于改变这种状况。Tasic 说,商业化对于推动单细胞测序至关重要,同样的情况也可能发生在空间技术中,我非常期待能够在单细胞水平上发挥作用的空间转录组学的商业解决方案。
10x Genomics 研发高级副总裁 Michael Schnall-Levin 说,空间技术,尤其是原位技术,在神经科学领域可能最先起飞。其他渴望空间分析的领域是发育生物学,人们研究细胞中分子的梯度和空间模式,以及癌症研究,尤其是评估肿瘤异质性和 T 细胞浸润。Schnall-Levin 表示,自公司成立以来,团队开始使用微流体技术、长读长测序方法,并专注于单细胞和空间分析。最初与 Spatial Transcriptomics 达成的潜在联合营销协议在 2018 年变成了一项收购。Visium 还达不到单细胞分辨率,但该公司正在努力实现这一目标。Schnall-Levin 说,他和他的团队还专注于基于原位的高分辨率分析。该公司收购了哈佛医学院 Church 实验室的衍生产品 ReadCoor 和斯德哥尔摩大学实验室和生命科学实验室研究员 Mats Nilsson 的衍生产品 Cartana。Cartana 使用挂锁探针与目标基因的 mRNA 杂交,然后进行滚环PCR以原位扩增 RNA,为测序做准备。ReadCoor 也使用条形码和探针进行荧光原位 RNA 测序和蛋白质检测。Schnall-Levin 说,Cartana 为 10x Genomics 带来了知识产权组合和具有专业知识的科学家,这是一种可以加速研究的资源。ReadCoor 带来了科学家、一项“基础技术”、知识产权和技术开发,因为制造仪器公司的团队已经克服了一些“非平凡”的工程限制。10x Genomics 的顾问 Shernaz Daver 说,在两家公司之间,10x 获得了 110 项专利,导致公司总共拥有 935 项专利。Schnall-Levin 说,这些技术能够将分子精确定位到亚细胞位置。目前,他们测量了几百个基因的集合,但显然,它们还有增长的空间。他认为 ReadCoor 和 Cartana 的方法是对支持 Visium 的技术的补充。该公司在收购后与学术实验室保持联系,因为他们拥有很重要的专业知识。
2019 年,nanoString 开始销售其用于空间分辨组织分析的仪器 GeoMx。该设备一直在与早期采用者进行测试。一些实验室使用原型,另一些实验室将样品发送到公司的西雅图总部并远程使用该仪器。“我们从中学到了很多东西,”首席科学官兼研发高级副总裁 Joe Beechem 说,“他们端着一杯酒坐在客厅里,控制着它,像小孩子一样大笑。”早期,GeoMx 可以捕获大约 100 个 mRNA;nanoString 总裁兼首席执行官 Brad Gray 说,现在这个数字已经超过 22,000,这表明该领域的发展速很快。nanoString 作为西雅图系统生物学研究所的衍生产品,拥有光学分子条码技术是关于空间分析的。Beechem 说,GeoMx 平台的早期使用者主要是在免疫肿瘤学领域,例如,实验室试图观察 T 细胞是否被困在肿瘤的外围,而不是将其破坏后深入其中。一天,就在演讲之前,他在餐巾纸上草草写下了该公司的寡核苷酸条码如何在空间上解析组织并在第二代测序仪上提供读数,该公司此前曾开发出一种将条形码附加到带有光可切割接头的抗体的方法,他的想法是在组织载玻片上散布连接器和条形码,寡核苷酸与 mRNA 杂交,显微镜拍摄组织快照,然后发出一束光,从光切割的接头上弹出,然后对 mRNA 和条形码进行测序。正如 Gray 解释的那样,他们的客户长期以来一直要求研究整个转录组并获得更高分辨率到细胞内亚细胞器的方法,以便他们可以问:“它是在表面上,还是在细胞核中,还是附着在某些细胞器上?”在他看来,市场将分为两个仪器类别。其一是诸如 GeoMx 和 Visium 之类的分析器,它们以高通量查看感兴趣的多细胞区域,然后是具有细胞和亚细胞分辨率的仪器,它们可能涵盖也可能不涵盖整个转录组,但每一次涵盖数千个基因。nanoString 刚刚推出了具有单细胞分辨率的 GeoMx 数字空间剖面仪。Gray 知道 nanoString 的竞争对手包括 Fluidigm 的 Hyperion 成像系统、成像质量细胞计数器和 Ionpath 的 MIBIscope。新的 nanoString 技术还使用光可切割标签对组织进行基因表达分析,包括福尔马林固定的石蜡包埋组织。例如,它被用于研究死于 COVID-19 的人的尸检样本的空间分析方面。Beechem 说,有了这种类型的技术,研究人员将拥有他们过去拥有的所有分子工具的所有功能。
BGI Research 正在开发一种用于空间分辨基因表达分析的高分辨率方法。BGI 集团首席执行官、BGI Research 负责人 Xun Xu 说,这个想法是弥合单细胞测序和细胞在组织中所处位置的空间分析之间的差距,这两者将共同塑造成功能和结构的研究。他观察了该领域的其他公司,并相信 BGI 方法将具有最高分辨率。Xu说,该方法可以捕获每个细胞数百个数据点,它作为纳米阵列封装在硅芯片上,采用半导体行业的光刻技术构建而成。BGI 已经使用这种方法来分析 DNA-蛋白质相互作用,BGI Research 的科学家 Chris Ao Chen 说,他和他的同事现在正在研究将细胞或组织嵌入这些芯片的方法,以产生高分辨率的空间解析数据。硅芯片阵列的隔间每个都有圆形区域,每个区域都包含可通过传统宽场成像解析的条形码 DNA 纳米球。DNA 纳米球是由捕获的转录本制成的环状 cDNA 束,包括准备用于 BGI 基于阵列的测序的引物和接头。每个条码读数都将阵列上的特定间距作为其空间地址。Xu说,芯片上的追踪线给出了在每个细胞中测量的坐标。空间方法BGI 借鉴了从 Complete Genomics, Inc. 获得的技术,预计将于 2021 年某个时候发布。BGI 已经制造出用于小鼠大脑空间分析的芯片,每个纳米球箱中约有 2,000 个基因和 4,000 个基因转录本。Xu说,该团队正在考虑从 mRNA 到其他组学。最终,该仪器可以帮助研究人员构建人类和模型生物参考组织图谱;他们可能会使用这种空间技术来评估遗传变异如何影响组织功能,例如在癌症中,或者将其用于跨物种比较,例如基于组织的进化和器官结构适应性研究。
涉及空间领域的公司还有活跃的软件开发商。Schnall-Levin 表示,信息学一直是公司的核心部分,他的团队致力于开源软件。研究人员可以将他们的算法和工具添加到 10x Genomics 流程中。正如Xu解释的那样,该团队致力于开发交互式可视化工具,让人们能够真实地了解组织。nanoString 的 Gray 说:“Joe 最大的研发部门现在是软件部门。”该公司的程序员比分子生物学家和工程师还多。
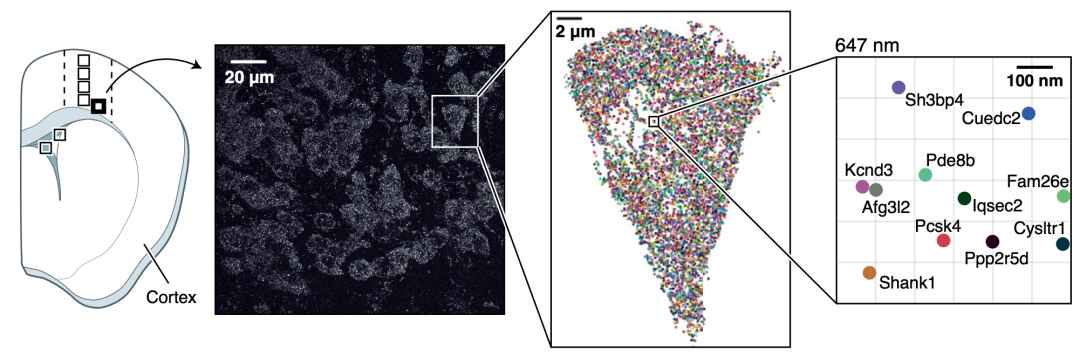
使用 MERFISH,Zhuang实验室捕获了人类癌细胞单个细胞中 10,050 个基因的表达。来自不同基因的 RNA 分子以不同的颜色显示。Credit: X. Zhuang laboratory, Harvard U./HHMI
6
Atlas at hand
空间分析对于 Brain Initiative 细胞普查网络 (BICCN) 至关重要,这是一个由美国国立卫生研究院资助的项目,其中许多实验室共同合作以制作人类、小鼠和非人类灵长类动物脑图谱。它涉及成像、电生理学和分子遗传学分析,如转录组学和表观基因组学。Allen Mouse Brain Atlas 的工作始于所有小鼠基因的原位表达谱,以显示解剖和空间基因表达模式,这是一个被广泛使用的参考数据库。Tasic 说,该团队正在小鼠和人脑中构建细胞类型的“周期性系统”。她领导了一个 BICCN 项目,该项目涉及用于全脑标记的细胞类型特定工具以及以细胞类型特定方式研究电路的方法。研究界很快将拥有特定于细胞类型的参考图谱,他们可以将自己的发现映射到该图谱上。在其他论文中,BICCN 团队最近发表了哺乳动物初级运动皮层的多模态细胞普查和图谱。Zeng 说,BICCN 第二阶段正在开始。在这些新项目中,有一些致力于细胞类型靶向工具,以应用空间和分子信息、扰乱细胞并表征它们在通路和行为中的作用。

艾伦脑科学研究所所长Hongkui Zeng(左)和同样在艾伦研究所的 Bosiljka Tasic。Credit: Allen Institute for Brain Science
对于最近的小鼠初级运动皮层图谱,研究小组指出,单细胞基因组学已经应用广泛,包括神经科学,并促进了实验室从能够对表型进行分类和描述到具有机械和解释性分子遗传框架的转变。为了通过转录组学细胞类型绘制来自小鼠初级运动皮层的 300,000 多个细胞的空间组织图,他们选择了 Zhuang 的 MERFISH,即多重抗错荧光原位杂交。他们将 MERFISH 与其他方法(如 Patch-seq)相结合,其中细胞通过电生理学进行测量然后测序。他们分析并比较了小鼠、狨猴和人类转录组细胞类型,然后选择了超过 250 个基因进行成像,通过单细胞表达谱进行聚类,发现与通过单核和单细胞 RNA-seq 识别的聚类非常吻合。这让他们能够辨别细胞类型的空间分布,并获得对传统定义层进行细化的皮质层的视图。他们还注意到沿内侧-外侧和前后轴的空间分布。有时,关于大脑制图的分歧会爆发,但 Tasic 相信先进的技术将慢慢有助于解决这些分歧,这些分歧有时是基于使用不同方法的实验室。有了一个共同的坐标系,分歧就可以转移到关于功能的必要讨论。“通常一个基因不足以识别细胞类型,”Zeng说,“你有多个基因在多个不同的水平上表达,它们共同构成了一个细胞。”观察单个细胞中表达的数千个基因的能力“已经极大地改变了这个领域”。空间转录组学增加了丰富性,可能还没有数以千计的基因,这取决于方法,但它的想法是相同的:研究人员以空间定位的方式了解细胞的许多基因。
Zhuang 很高兴 MERFISH 被用于构建脑图谱。“我个人对大脑非常着迷,”她说。在 MERFISH 中,RNA 在杂交轮次中被编码。非荧光靶向探针与 mRNA 结合,荧光读出探针与靶向探针杂交,不同的条形码。多轮次的杂交、条形码和纠错使捕获许多基因成为可能。然而,杂交轮次的挑战是错误的积累,庄说。每一轮单独的错误率很小,但错误累积并变得显着,这有基因错误识别的风险。smFISH 具有很高的准确性,庄说。但它是一次性测量,而不是累积误差的连续测量。当她和她的团队开发 MERFISH 时,他们致力于成像、内置纠错以及一种在数千种 RNA 物种转录组范围内提供结果的方法。“这花了我们一段时间,”她说。该方法实现了80%的检测效率。在另一个基于 BICCN 的项目中,Zhuang和她的团队使用 MERFISH 分析了小鼠下丘脑视前区超过一百万个细胞。在以前没有生过幼崽的雌性小鼠的大脑中,接触幼崽会激活一个神经元,这种神经元在雌性和雄性小鼠父母中也被激活,但在没有生育过一胎的雄性小鼠中则不活跃。她和她的团队还应用 MERFISH 来表征亚细胞空间组织,以揭示不同细胞区室中的 RNA 以及染色质的组织方式。最终,这项工作展示了“3D 基因组组织和转录是如何相互关联的,”她说。与哈佛医学院研究员大卫沃尔特和杰弗里莫菲特(她的前博士后研究员现在拥有自己的哈佛医学院实验室)一起,她成立了一家名为 Vizgen 的公司,将 MERFISH 商业化。
7
Future in space
作为一名显微镜专家,Zhuang很高兴看到成像和单细胞基因组分析的领域越来越近。KTH 的 Ståhl 期待高分辨率的空间分辨转录组学,“也许,像 10 微米这样的单个细胞可能是最佳点,”他说,它可以为每个像素提供足够的数据。Zeng 对出现的空间分析技术感到高兴。许多现有方法比如scRNAseq,它仍然为量化基因表达提供最高灵敏度的读数。不过,有一天,他希望我们可以完全摆脱单细胞测序以进行全转录组的单细胞空间分析。
空间信息显然很重要,Shendure 说,就人们可以想象的技术最终走向何方而言,我们仍处于早期阶段。令人兴奋的是,空间解析转录组学是年度最佳方法,Macosko 说,我认为这只是一个开始。为了深入研究一个感兴趣的问题,人们可能会使用空间分析领域的众多工具和技术中的几种。标准、指标和基准将有助于空间领域,就像它们有助于单细胞基因组学领域一样。
Macosko 说,从单分子 FISH 和单细胞基因组分析开始,已经成为将基因组学的大规模假设生成技术带入细胞生物学和组织生物学的一组方法——这将奠定思维和技术发展的基础。研究人员将能够在空间环境中对基因表达进行细致研究,并进行其他测量,例如酶促过程和细胞之间、基因之间和蛋白质之间的相互作用。他们将能够对空间中感兴趣的方面进行编码和锚定,这将改变细胞生物学以及病理学和组织学的实践方式。
Zeng看到了许多未来的机会。在很多情况下,这些技术并不完美,我们想要更多更好的东西,以扩大技术并能够常规有效地测量数千个基因。Tasic 说,空间方法将成为标准。总有一天人们会从组织取回空间解析的单细胞遗传信息,对该领域论文的常见反应可能会变成:“哦,你没有测量所有基因?为什么不?”