
基于Salmon的转录组定量流程
为什么使用Salmon?
Salmon是不基于比对计数而直接对基因进行定量的工具,适用于转录组、宏基因组等的分析。
其优势是:
定量时考虑到不同样品中基因长度的改变(比如不同isoform的使用)
速度快、需要的计算资源和存储资源小
敏感性高,不会丢弃匹配到多个基因同源区域的reads
可以直接校正GC-bias
自动判断文库类型
39个转录组分析工具,120种组合评估表明Salmon的定量准确性和稳定性都比较好。
其原理如下图所示,概括来讲是通过构建统计模型来推测已经注释的转录本呈现出什么表达模式时我们才会测序产生当前的FASTQ数据:
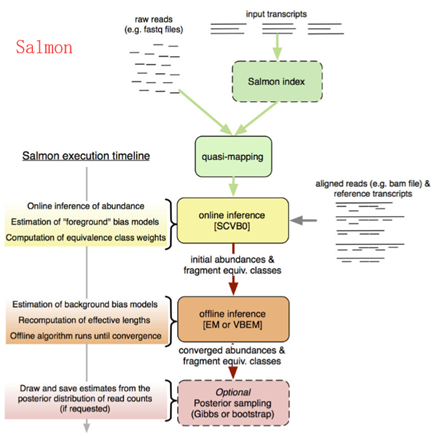
怎么使用Salmon?
Salmon定量依赖于cDNA序列和原始的FASTQ序列,新版本也可以提供基因组序列以处理某些能同时比对到已经注释的基因区和基因间区的reads,获得更准确地定量结果。
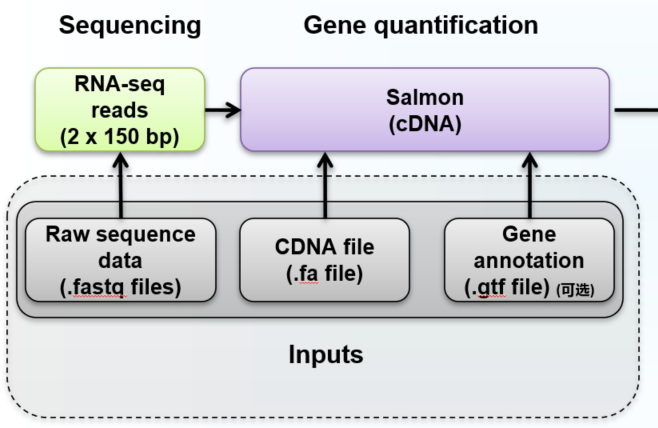
第一步,构建索引
从ENSEMBL下载基因组和基因注释文件,具体参考NGS基础 - 参考基因组和基因注释文件。
mkdir -p genome
cd genome
# GRCh38.fa 人基因组序列,从Ensembl下载
# GRCh38.gtf 人基因注释序列,从Ensembl下载
wget ftp://ftp.ensembl.org/pub/release-100/gtf/homo_sapiens/Homo_sapiens.GRCh38.100.gtf.gz -O GRCh38.fa.gz
wget ftp://ftp.ensembl.org/pub/release-100/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz -O GRCh38.gtf.gz
gunzip -c GRCh38.fa.gz >GRCh38.fagunzip -c GRCh38.gtf.gz >GRCh38.gtf# 获取cDNA序列
gffread GRCh38.gtf -g GRCh38.fa -w GRCh38.transcript.fa.tmp
# gffread生成的fasta文件同时包含基因名字和转录本名字
grep '>' GRCh38.transcript.fa.tmp | head
# 去掉空格后面的字符串,保证cDNA文件中fasta序列的名字简洁,不然后续会出错
cut -f 1 -d ' ' GRCh38.transcript.fa.tmp >GRCh38.transcript.fa
构建索引
# 获取所有基因组序列的名字存储于decoy中
grep '^>' GRCh38.fa | cut -d ' ' -f 1 | sed 's/^>//g' >GRCh38.decoys.txt
# 合并cDNA和基因组序列一起
# 注意:cDNA在前,基因组在后
cat GRCh38.transcript.fa GRCh38.fa >GRCh38_trans_genome.fa
# 构建索引 (更慢,结果会更准)
salmon index -t GRCh38_trans_genome.fa -d GRCh38.decoys.txt -i GRCh38.salmon_sa_index定量单样品FASTQ数据
cd ../
fastq-dump -v --split-3 --gzip SRR1039521
rename "SRR1039521" "trt_N061011" SRR1039521*
# -p: 表示若待创建的文件夹已存在则跳过;若不存在,则创建;也可用于创建多层文件夹
# man mkdir 可查看详细帮助
mkdir -p trt_N061011
# -l: 自动判断文库类型,尤其适用于链特异性文库
# The library type -l should be specified on the command line
# before the read files (i.e. the parameters to -1 and -2, or -r).
# This is because the contents of the library type flag is used to determine how the reads should be interpreted.
# --gcBias: 校正测序片段GC含量,获得更准确的转录本定量结果
# One can simply run Salmon with --gcBias in any case,
# as it does not impair quantification for samples without GC bias,
# it just takes a few more minutes per sample.
# For samples with moderate to high GC bias, correction for this bias at the
# fragment level has been shown to reduce isoform quantification errors
salmon quant --gcBias -l A -1 trt_N061011_1.fq.gz -2 trt_N061011_2.fq.gz -i genome/GRCh38.salmon_sa_index -g genome/GRCh38.gtf -o trt_N061011/trt_N061011.salmon.count -p 10定量后输出结果存储于trt_N061011/trt_N061011.salmon.count目录中
# 输出结果存储在 trt_N061011/trt_N061011.salmon.count目录中
# quant.sf 为转录本表达定量结果,第4列为TPM结果,第5列为reads count
# quant.genes.sf 为基因表达定量结果
head -n 30 trt_N061011/trt_N061011.salmon.count/quant.sf | tail定量结果为
Name Length EffectiveLength TPM NumReads
ENST00000609179 1196 1052.656 0.000000 0.000
ENST00000492242 1277 1058.126 92.111195 19.918
ENST00000382291 2088 1963.212 1447.695765 580.820
ENST00000382285 1329 1183.099 211.657526 51.174多样品定量
未完待续
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集
(请备注姓名-学校/企业-职务等)
评论