单细胞转录组基本概念(一)
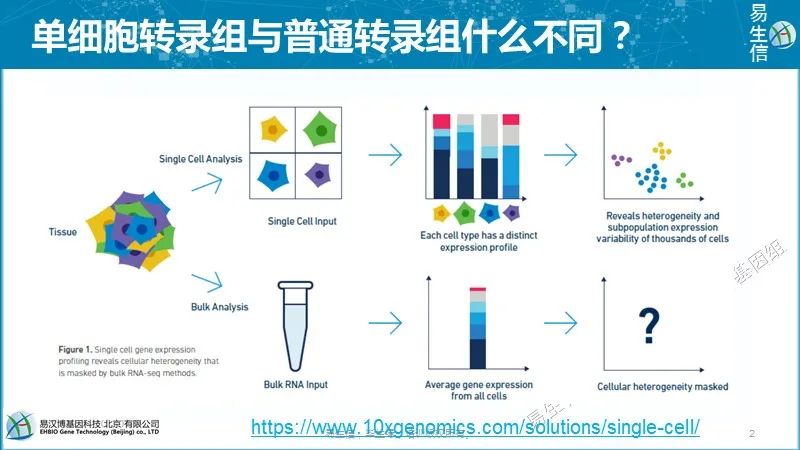
普通转录组的思路也可以应用到单细胞转录组。普通转录组相当于把一群细胞或一个器官混合到一起去提取RNA,获得的是每个细胞中RNA表达量的平均值。单细胞是把每个细胞单独分出来去提取RNA,然后建库测序,获得是是单个细胞的表达值。在每个细胞里面基因的表达具有随机性,且存在异质性。而且这些细胞群中会存在不同类型的细胞,尤其是当我们对整个组织或者器官进行测序时,它们本身就是由不同类型的细胞组成的,而我们用普通转录组来测序,相当于掩盖住了这些不同的细胞类型的差异,展示的是整个组织的平均的状态,所以说单细胞从这个来看跟普通转录组就不同在是用一个细胞测,不是用一堆细胞测。
 既然是做单细胞,第一步就是把单个细胞分选出来。分选的方式也比较多,物理切割,酶消化,FACS分选等。假如已经分到了一个单细胞,跟常规的转录组步骤上是一样的。下一步就是把单细胞里的RNA提取出来去建库测序。但是一个细胞里的RNA含量是比较少的,难建库成功。这个里面“量”的概念很有意思。比如说单细胞量少,需要特殊处理。到我们做计算的,量大需要特殊处理。比如微信这个工具,看上去谁都可以做,就是一个信息发送和接收的工具,技术难度不大。但如果是做成承载8亿用户的平台,难度就大了很多了。后面讲到的单细胞聚类也是,数量一大,就得先降维再聚类。建库之前需要做一步扩增,扩增主要有2个方式,一个是体外转录,另一个是常规的PCR扩增。因为单细胞的RNA量比较少,所以要扩增的话循环数就会比较多,会引入一些PCR扩增带来的偏好性,为了解决这个问题,就有了现在的第2个技术——
既然是做单细胞,第一步就是把单个细胞分选出来。分选的方式也比较多,物理切割,酶消化,FACS分选等。假如已经分到了一个单细胞,跟常规的转录组步骤上是一样的。下一步就是把单细胞里的RNA提取出来去建库测序。但是一个细胞里的RNA含量是比较少的,难建库成功。这个里面“量”的概念很有意思。比如说单细胞量少,需要特殊处理。到我们做计算的,量大需要特殊处理。比如微信这个工具,看上去谁都可以做,就是一个信息发送和接收的工具,技术难度不大。但如果是做成承载8亿用户的平台,难度就大了很多了。后面讲到的单细胞聚类也是,数量一大,就得先降维再聚类。建库之前需要做一步扩增,扩增主要有2个方式,一个是体外转录,另一个是常规的PCR扩增。因为单细胞的RNA量比较少,所以要扩增的话循环数就会比较多,会引入一些PCR扩增带来的偏好性,为了解决这个问题,就有了现在的第2个技术——体外转录。这个技术不通过扩增,在RNA上加一个T7的启动子,让它一轮一轮的转录出新的RNA,实现线性扩增。
PCR也有对应的线性扩增的技术,当获得了可以反转录成足够量cDNA的RNA之后,就可以把它按照之前常规转录组的方式去建库测序,后续的分析也是比较类似。
因为单细胞里面RNA的量少,所以说整个富集的过程中会出现一部分基因在一个细胞里能检测到,在另外一个细胞里面检测不到,而每个细胞里面的检测存在一个随机性,同时单细胞测序深度比较低,所以说分析时相比于普通转录组有一些是需要特别注意,但整体的分析思路是类似的。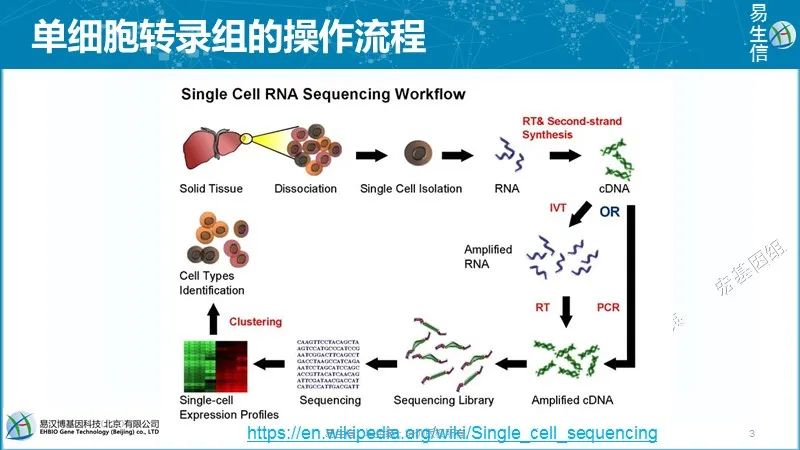 单细胞转录组的分析流程,主要还在在后期的聚类、发育轨迹、整合分析等。
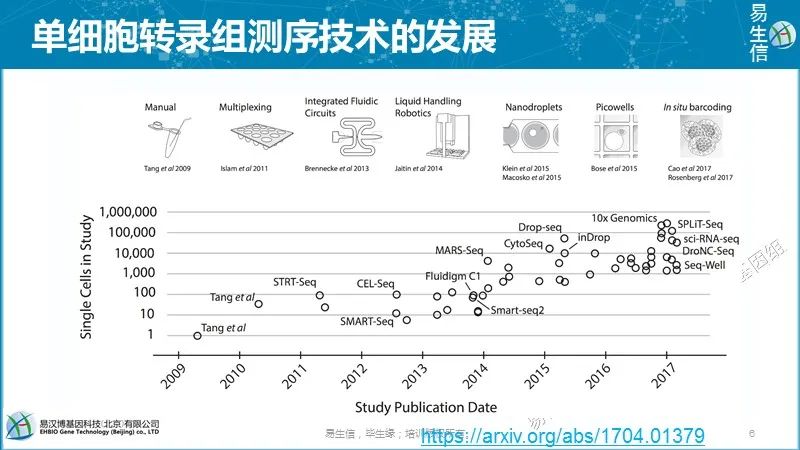
单细胞转录组的分析流程,主要还在在后期的聚类、发育轨迹、整合分析等。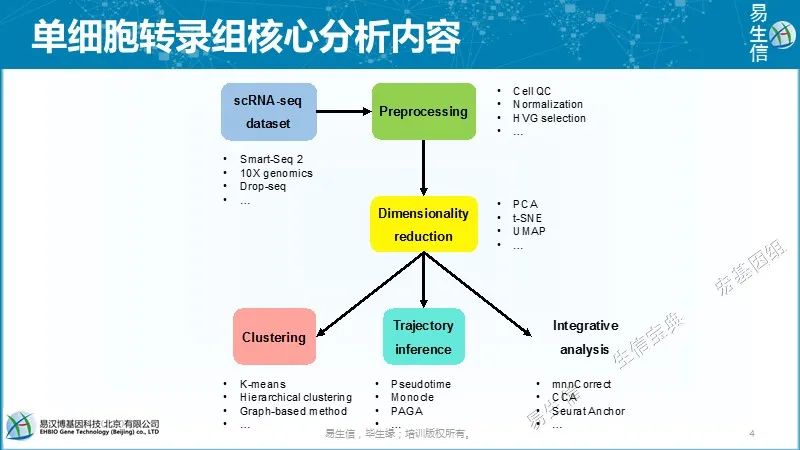 下图是单细胞转录组测序技术的发展,横轴是时间,纵轴是每一个技术所能检测到的细胞的量的变化,基本服从指数的分布。
下图是单细胞转录组测序技术的发展,横轴是时间,纵轴是每一个技术所能检测到的细胞的量的变化,基本服从指数的分布。
1992年Eberwine教授采用体内反转和体外转录技术测定了单个细胞里的数个基因的表达。后续非靶向的mRNA扩增技术的发展为2009年汤富酬老师打响单细胞测序前两枪提供了技术基础。
汤富酬老师在国外做博后的时候,2009、2010年2篇文章拉开了单细胞转录组测序的序幕,现在他也是单细胞领域特别高产的研究者。当时单细胞转录组测序主要应用于特别难获取的细胞,比如说胚胎发育早期,合子,二细胞,四细胞,八细胞期,这时候因为每一个阶段细胞的数目都是很有限的,当时就想着能够开发一个技术对这种含量特别少的细胞能够提取建库成功,然后获得它们的表达量,从而来研究这些常规转录组所研究不了的生物过程,所以当时的发展是尽量提高测序的深度。
到了后来也还是2010年,也是目前在单细胞领域比较火的一个老师,郭国骥老师,他在哈佛做博后时用fluidigm的一个设备检测了500个细胞的48个基因的单细胞的RT-qPCR结果,发现用这48个基因可以对500个细胞进行很好的细胞分型,定义每个细胞的类型。所以大家看到这篇文章开始逐渐意识到,完全可以在单细胞分析上以量取胜,就是每个细胞可以测的比较浅,但是测很多细胞,这样对鉴定细胞类型很有帮助,所以说后续技术的大部分优化点都在于如何提高检测通量上,而现在我们已经可以检测几千或者上万的细胞。
后续就由此发展出来很多技术,比如drop-seq,indrop,10Xgenomics,这些都是基于droplets的技术。早期单细胞的分选主要靠人工,用移液管,移液枪或者显微操作去把细胞单个单个的分出来,再放到微孔里一个一个进行反应,或者使用fluidigm的微流控设备或者操作机器人,之后就有了更自动化的设备,使得我们用更低的成本,更少的时间来检测出来更多的细胞。
这些技术都不能保留细胞原始的空间上的位置,而In situ barcoding或者Picowells可以让我们得知这个细胞在原始空间上谁跟谁更近,同时可以检测出来这些细胞里面基因的表达量,提供另外一个维度的信息。
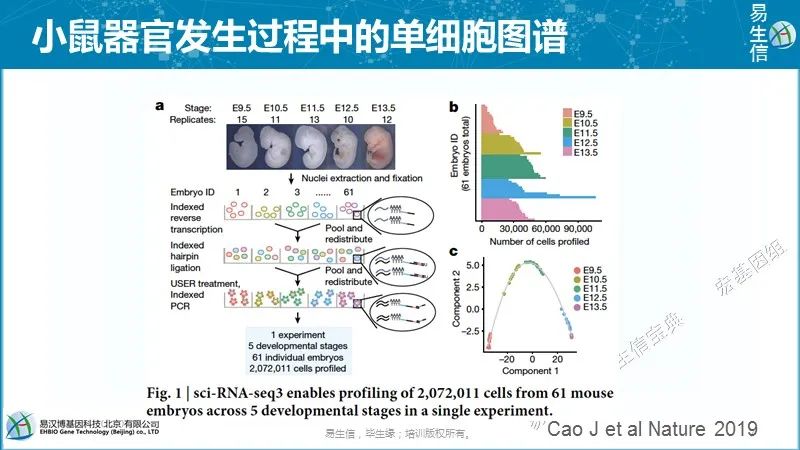
郭国骥老师另外一篇cell中的Microwell-seq可以检测数万个细胞,是属于测序成本比较低的技术,后面再讲它的基本应用。
原位序列条形码标记(例如单细胞组合索引RNA测序(sci-RNA-seq)和基于分池连接的转录组测序(split-poolligation-based transcriptomesequencing, SPLiT-seq)
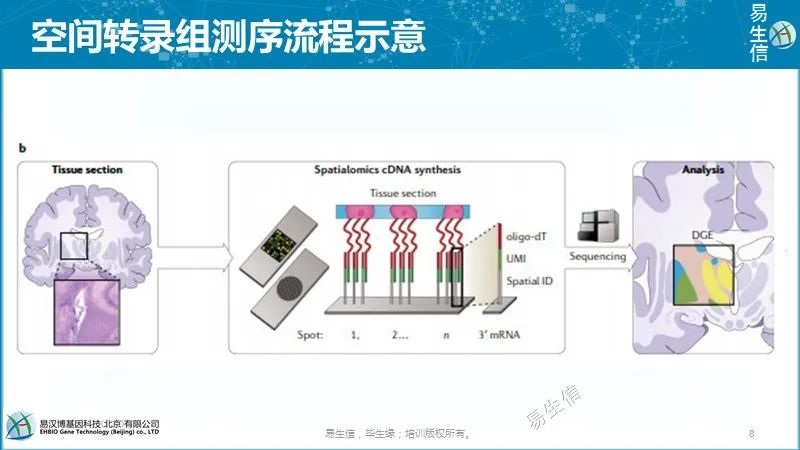
 在SpatialTranscriptomics(美国10XGenomics公司)和Slide-seq方法中,采用寡核苷酸芯片(oligo-arrayed microarray slides)和布满寡核苷酸的凝珠 (denselypacked oligo-coatedbeads) 直接从冷冻组织切片中捕获RNA进行测序。寡核苷酸包含spatialbarcode,UMI和oligo-dT引物,可唯一识别每个转录本及其位置。测序reads比对回玻片坐标获得空间基因表达信息。
在SpatialTranscriptomics(美国10XGenomics公司)和Slide-seq方法中,采用寡核苷酸芯片(oligo-arrayed microarray slides)和布满寡核苷酸的凝珠 (denselypacked oligo-coatedbeads) 直接从冷冻组织切片中捕获RNA进行测序。寡核苷酸包含spatialbarcode,UMI和oligo-dT引物,可唯一识别每个转录本及其位置。测序reads比对回玻片坐标获得空间基因表达信息。
已经证明,SpatialTranscriptomics可用于多种物种的组织,包括小鼠脑和人乳腺癌组织、人心脏组织和拟南芥花序组织。Slide-seq是一项最新开发的技术,已显示可用于小鼠大脑的冷冻切片分析。这些直接的mRNA捕获方法不需要专门的设备,具有相对简单的分析方法,并且可能大规模应用于许多组织。
但是,有两个重要的问题有待解决。首先,该技术只能应用于新鲜的冷冻组织。其次,分辨率受到芯片大小和寡核苷酸凝珠间距的限制;当前应用的芯片大小分别为6.5×7mm和3×3mm,限制了可以检测的组织切片的大小。SpatialTranscriptomics的凝珠直径为100µm,间隔为100µm,这意味着它们不够小或不够密,以致无法实现单细胞分辨率。Slide-seq的凝珠 (beads)小得多,直径仅为10 μm,并且堆积致密,提供了十倍的空间分辨率,大约一半的beads可以获得单个细胞数据。
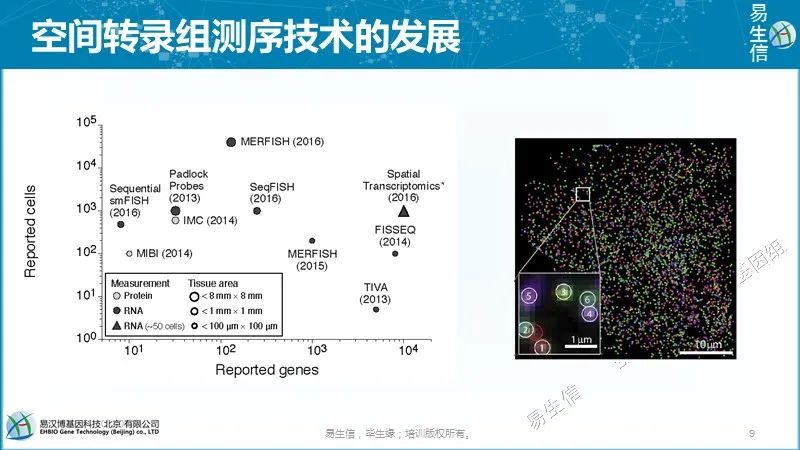
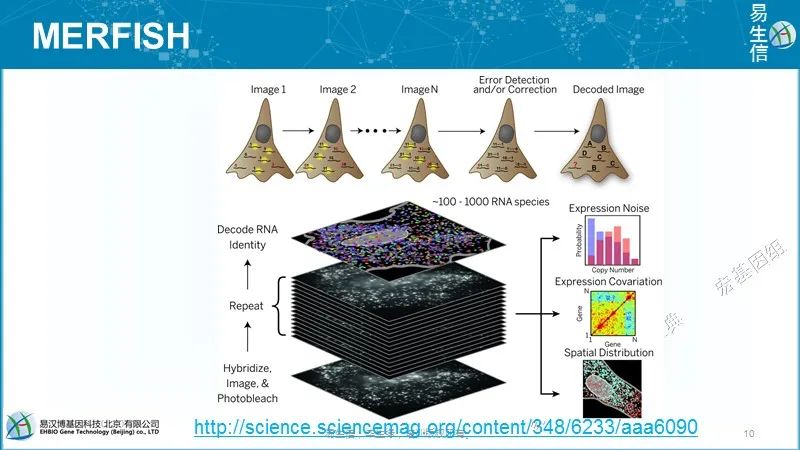 真正想了解大脑,你还需要一个空间背景(spatial context),因为大脑细胞不像肝脏或其他器官那样以对称的方式组织,大脑的不同寻常之处在于它具有神经元的拓扑结构。因此,我们希望能够观察大脑的一部分,看看那里有哪些细胞、它们在哪里,以及它们周围有哪些类型的细胞。
真正想了解大脑,你还需要一个空间背景(spatial context),因为大脑细胞不像肝脏或其他器官那样以对称的方式组织,大脑的不同寻常之处在于它具有神经元的拓扑结构。因此,我们希望能够观察大脑的一部分,看看那里有哪些细胞、它们在哪里,以及它们周围有哪些类型的细胞。
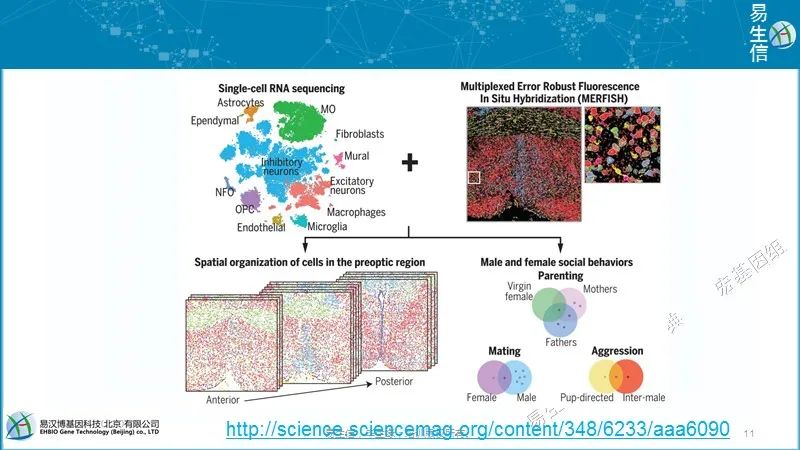
MERFISH的主要应用之一是原位识别细胞类型。不同的细胞类型有不同的基因表达谱。因此,这些基因表达谱为细胞类型鉴定提供了定量和系统的方法。由于我们可以通过MERFISH成像在完整组织中做到这一点,我们也可提供这些细胞类型的空间结构(spatial organization)。
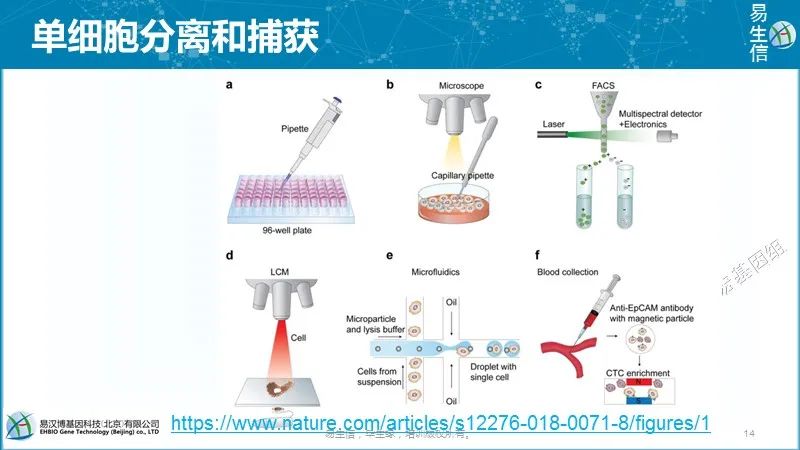
 极限稀释加移液枪分离单细胞;显微操作分选单细胞;流式分选带有表面Marker的单细胞;激光切割实体组织;微流控技术;磁珠捕获,主要用于CTC
极限稀释加移液枪分离单细胞;显微操作分选单细胞;流式分选带有表面Marker的单细胞;激光切割实体组织;微流控技术;磁珠捕获,主要用于CTC
 它的一个优点是可以结合流式细胞荧光分选(FACS, fluorescent activated cell sorting)根据表面Marker分选细胞。因此特别适合分选细胞子集用于测序。它的另一个优点是可以获得细胞形态全览图,提供另外一个维度的信息,可用于鉴定微孔中是否有损伤的细胞或双份细胞,主要缺点是通量低且每个细胞所需的工作量相当大。
它的一个优点是可以结合流式细胞荧光分选(FACS, fluorescent activated cell sorting)根据表面Marker分选细胞。因此特别适合分选细胞子集用于测序。它的另一个优点是可以获得细胞形态全览图,提供另外一个维度的信息,可用于鉴定微孔中是否有损伤的细胞或双份细胞,主要缺点是通量低且每个细胞所需的工作量相当大。 微流型平台,比如Fluidigm’s C1,提供了一个更加整合的系统,同时可以捕获细胞和完成文库构建的准备过程。比微孔型平台通量更高,但只能捕获10%的细胞,不适合处理稀有细胞或细胞量量很少的情况。
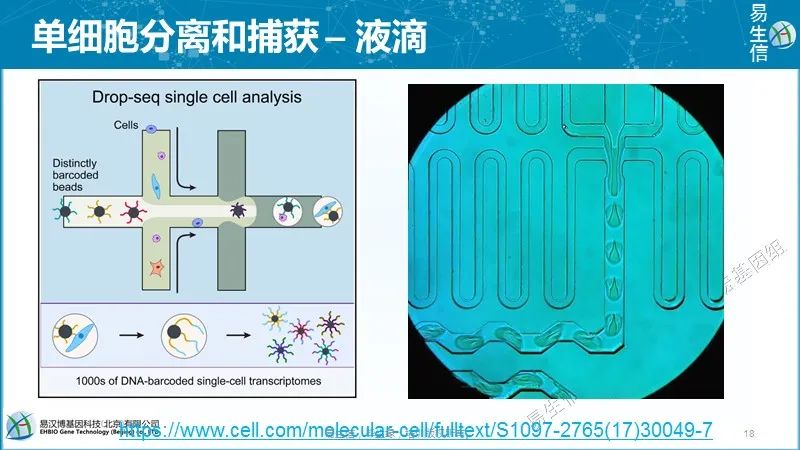
微流型平台,比如Fluidigm’s C1,提供了一个更加整合的系统,同时可以捕获细胞和完成文库构建的准备过程。比微孔型平台通量更高,但只能捕获10%的细胞,不适合处理稀有细胞或细胞量量很少的情况。 液滴型方法是将单独的细胞和一个包含建库所学酶的珠粒(bead)包裹在一个纳米级液滴里面。特殊地,每个珠粒(bead)包含一段独特的条形码序列(barcode),会加到所有来自于液滴里面这个细胞的序列上,用于区分不同细胞的转录本。
液滴型方法是将单独的细胞和一个包含建库所学酶的珠粒(bead)包裹在一个纳米级液滴里面。特殊地,每个珠粒(bead)包含一段独特的条形码序列(barcode),会加到所有来自于液滴里面这个细胞的序列上,用于区分不同细胞的转录本。
 采用光刻技术制作微孔矩阵硅片(微孔直径28 um,深度35 um,100,000个微孔),以此为模具制作PDMS微柱模具。这两个模具可以反复使用。最终用于富集的微孔板是通过倾到5%的琼脂糖凝胶到PDMS微柱模具上生成的。细胞悬液加到凝胶微孔模具上,利用重力使细胞落入微孔,通常一个微孔只能容纳一个细胞,一块板子可以同时捕获约10000个单细胞。每一步操作都可视、可控制,doublets可以通过镜检洗除。随后每个孔加入包含107-108特定探针集的与孔径大小匹配的磁珠,标记每个细胞中的mRNA(每个磁珠的寡核苷酸序列中都有一段特异的序列用于标记细胞来源),然后使用Smart-seq2方法进行后续的反转录、扩增。扩增后的cDNA片段使用转座酶片段化(这步倒有些类似ATAC-seq),富集3’末端转录本序列测序。
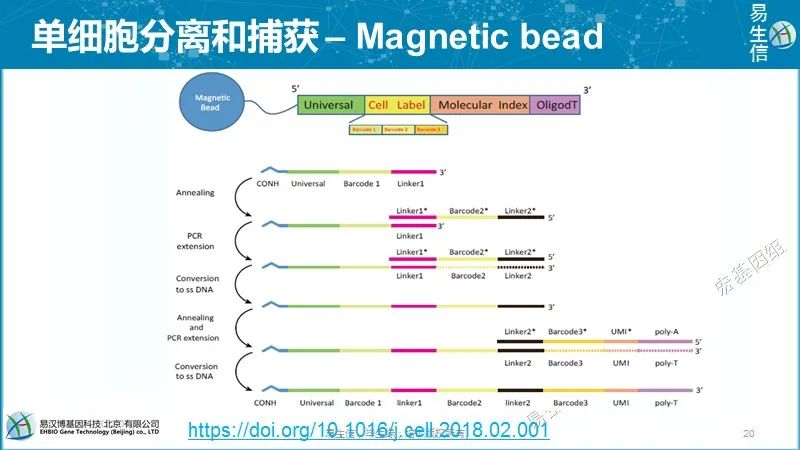
采用光刻技术制作微孔矩阵硅片(微孔直径28 um,深度35 um,100,000个微孔),以此为模具制作PDMS微柱模具。这两个模具可以反复使用。最终用于富集的微孔板是通过倾到5%的琼脂糖凝胶到PDMS微柱模具上生成的。细胞悬液加到凝胶微孔模具上,利用重力使细胞落入微孔,通常一个微孔只能容纳一个细胞,一块板子可以同时捕获约10000个单细胞。每一步操作都可视、可控制,doublets可以通过镜检洗除。随后每个孔加入包含107-108特定探针集的与孔径大小匹配的磁珠,标记每个细胞中的mRNA(每个磁珠的寡核苷酸序列中都有一段特异的序列用于标记细胞来源),然后使用Smart-seq2方法进行后续的反转录、扩增。扩增后的cDNA片段使用转座酶片段化(这步倒有些类似ATAC-seq),富集3’末端转录本序列测序。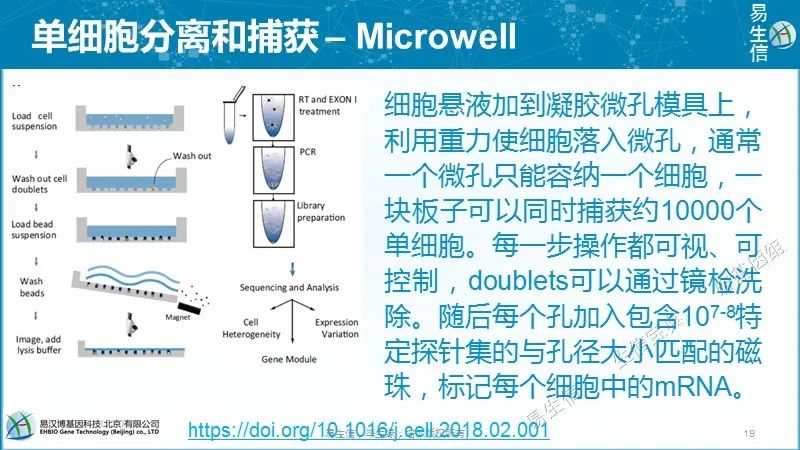

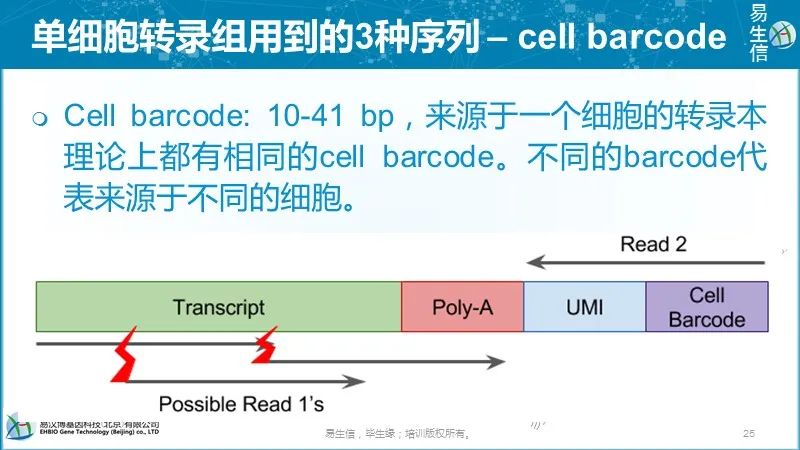
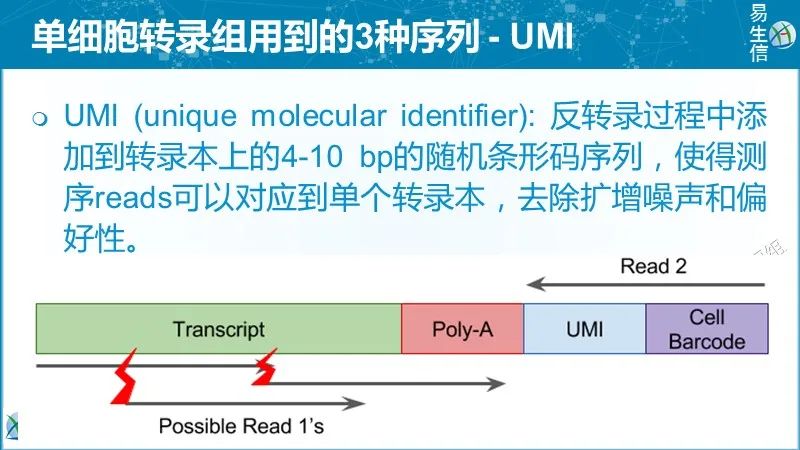
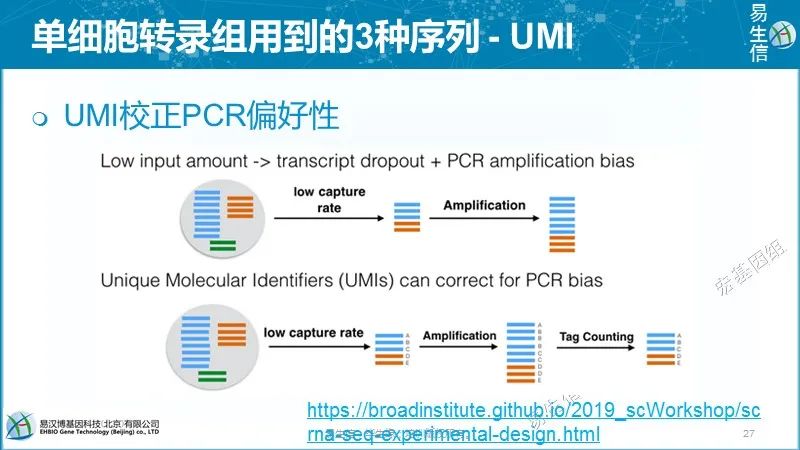
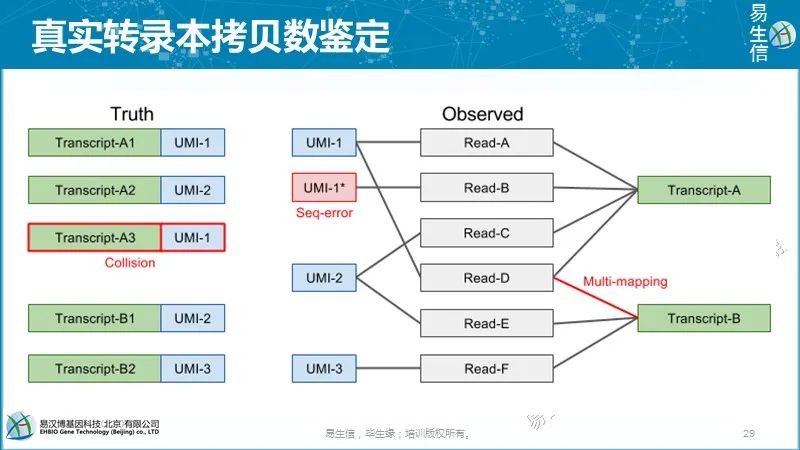
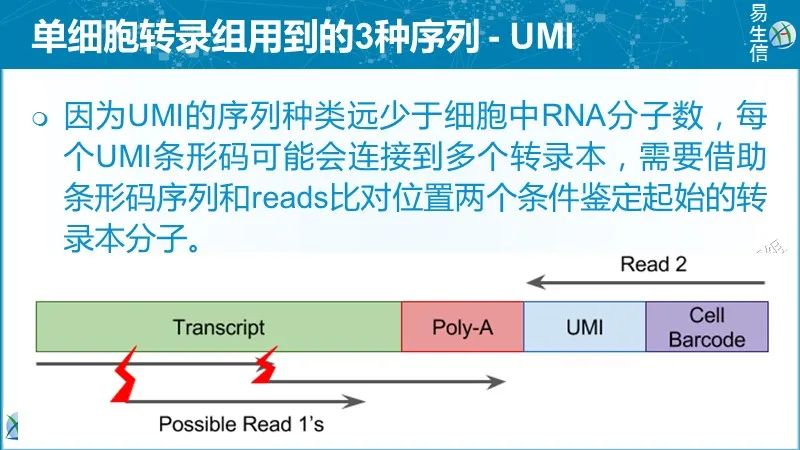 理论上,每个唯一的UMI-转录本对应该对应来源于一个RNA分子的所有reads。但是现实往往并非如此,最常见的原因是:
理论上,每个唯一的UMI-转录本对应该对应来源于一个RNA分子的所有reads。但是现实往往并非如此,最常见的原因是:
不同的UMI序列不一定表示它们是不同的UMI分子 由于PCR或测序错误,碱基替换可能导致新的UMI序列。较长的UMI出现碱基替换的机会更多。根据细胞条码测序错误估计,7-10%的10 bp长度的UMI中至少有一个碱基替换。如果错误没有纠正,将会过高估计转录本的数目。
不同的转录本不一定是不同的分子 比对错误或多个比对位置可能导致某些UMI对应到错误的基因/转录本。这种类型的错误也会导致过高估计转录本的数目。
相同的UMI不一定意味着相同的分子UMI频次的不同和短UMI可导致同一UMI和相同基因的不同mRNA分子相连,进而可能低估转录本数量。

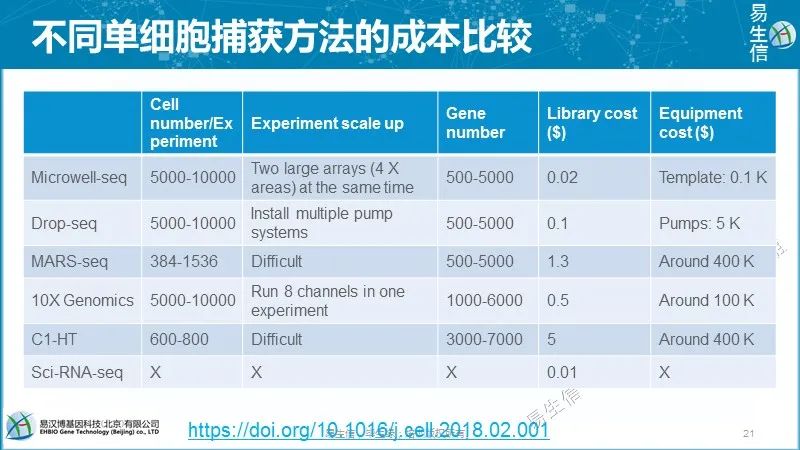
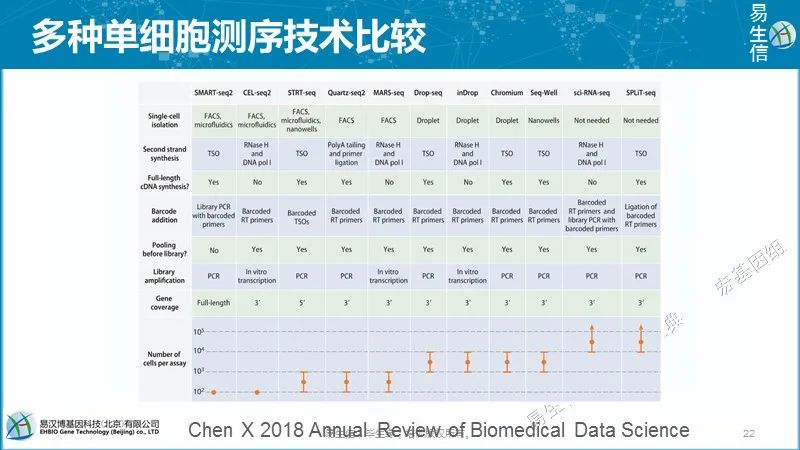
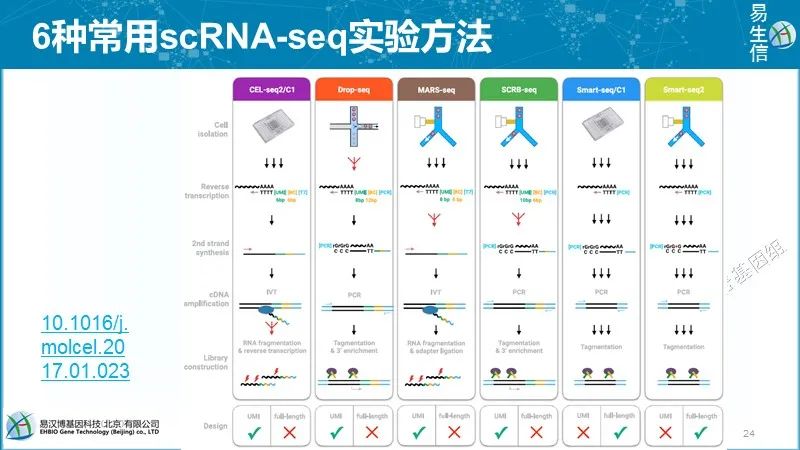
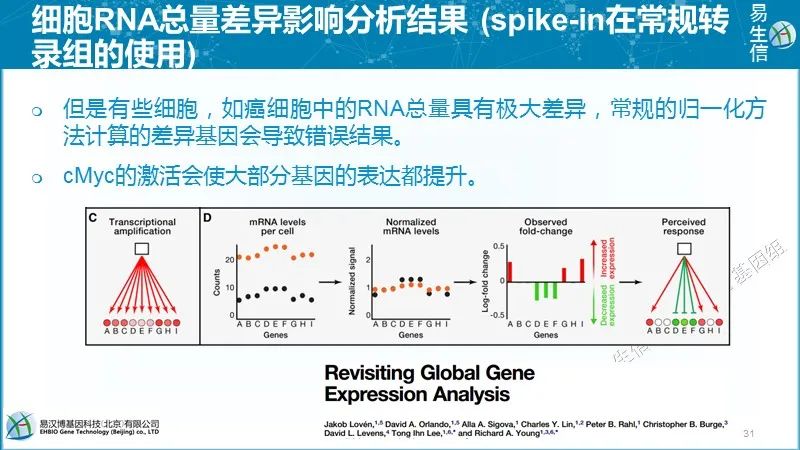 后面再继续不同技术之间的比较
后面再继续不同技术之间的比较
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集