Nature | 癌症中转录组变化的基因组基础 (1)
链接:doi.org/10.1038/s41586-020-1970-0
摘 要
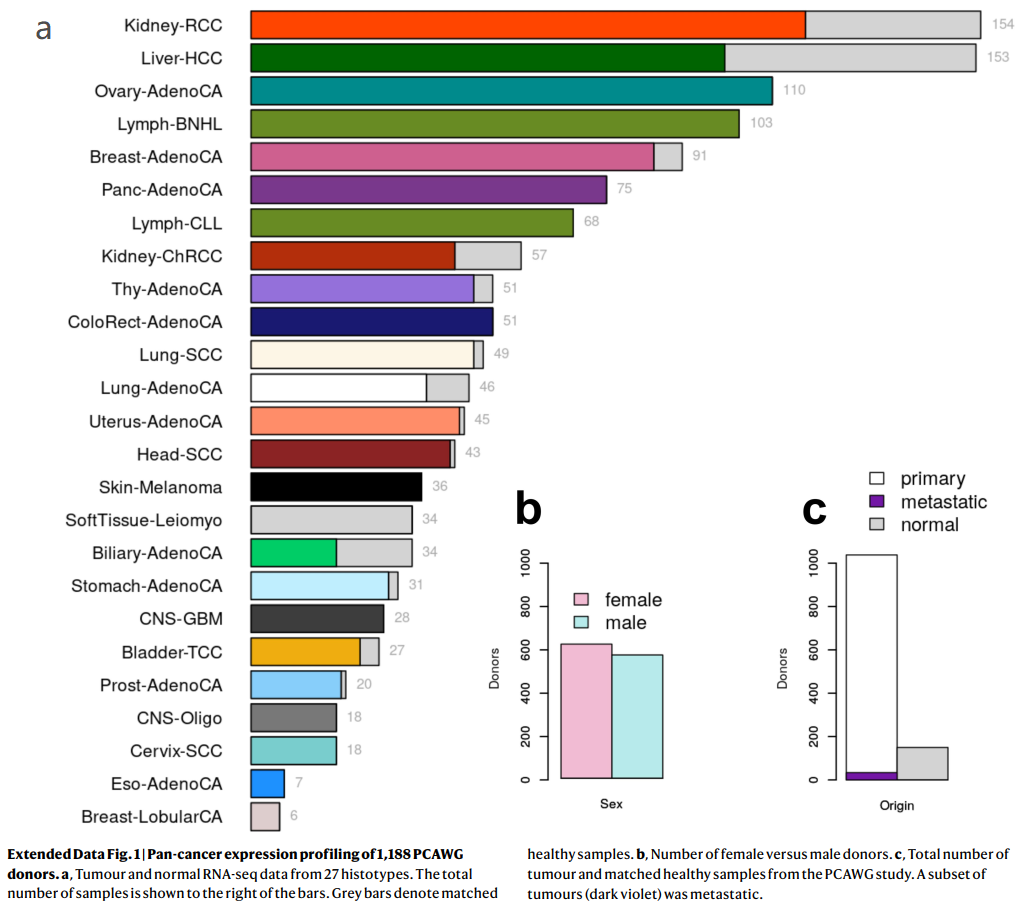
Extended Data Fig. 1 | 1,188例PCAWG捐献者的泛癌表达谱
a,来自27种组织类型的肿瘤和正常RNA-seq数据。样本总数显示在柱状图的右边。灰色条表示匹配的健康样本。
b,女性和男性捐献者的数量。
c,来自PCAWG研究的肿瘤总数和匹配的健康样本。一组肿瘤(深紫色)已转移。
癌症特异性胚系顺式-eQTLs
Cancer-specific germline cis-eQTLs
为了研究不同类型RNA改变的潜在机制,我们首先关注了基因表达水平的变化 (Extended Data Fig. 2)。
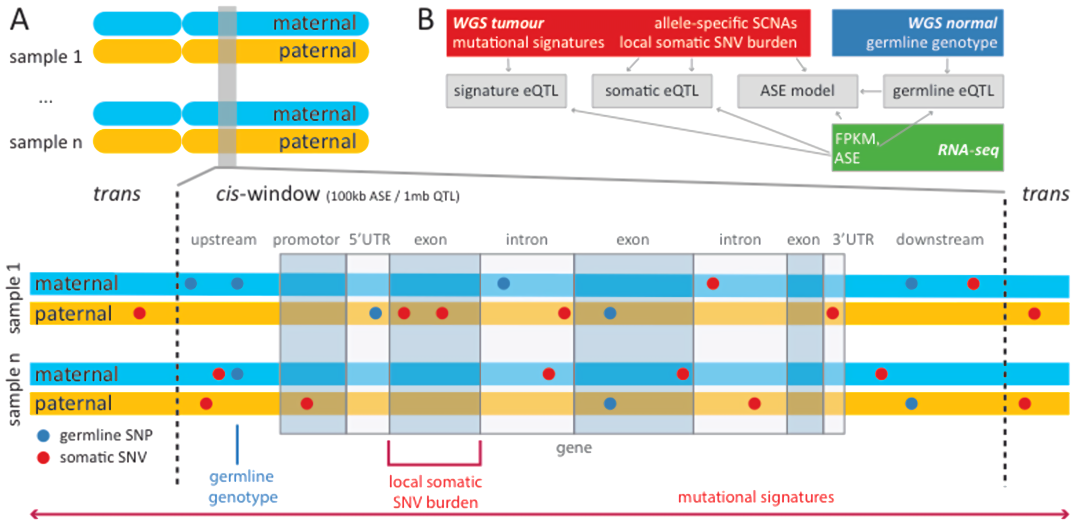
Extended Data Fig. 2 | 概述:在分析中考虑的遗传变异的不同来源
所有体细胞顺式eQTL分析的窗口大小为1 Mb;ASE与胚系顺式eQTL分析的窗口大小为100 kb。
b,概述:不同的数据集,及其对a中所述分析的贡献。
ASE和总表达 (Total expression/FPKM)来自肿瘤和正常RNA-seq数据。箭头表示所执行的单个分析之间的依赖关系。
我们最初考虑了常见的胚系变异 (次要等位基因频率 (Minor allele frequency, MAF)≥1%)靠近单个基因 (±100 kb),并在队列中绘制了表达定量性状位点 (eQTL) (Extended Data Fig. 3, Supplementary Table 1)。
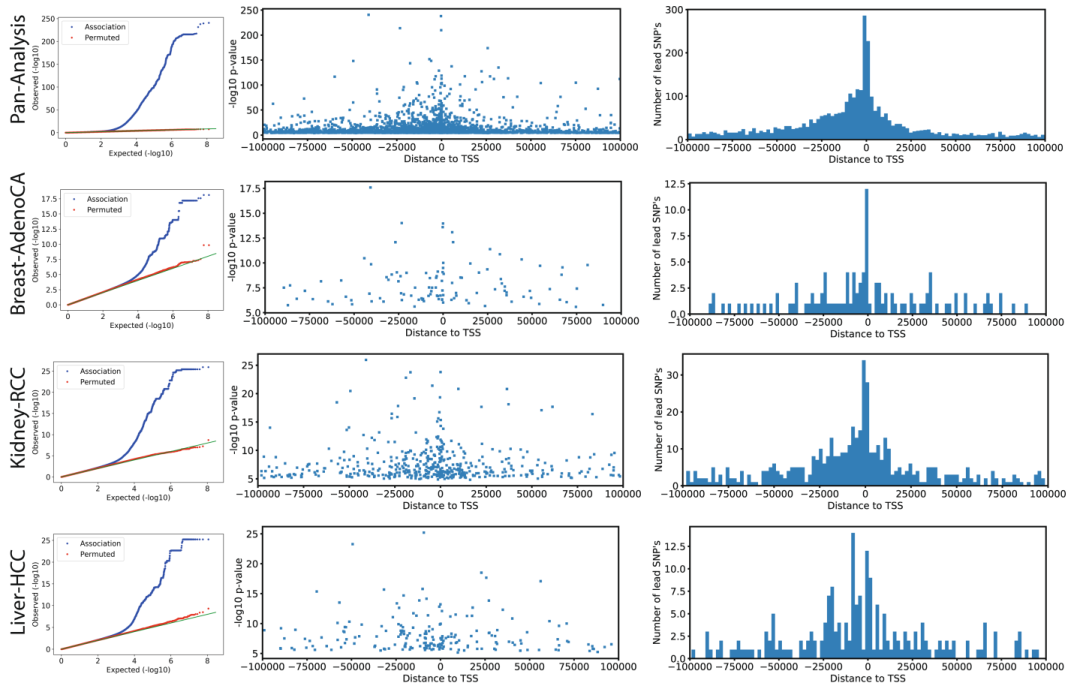
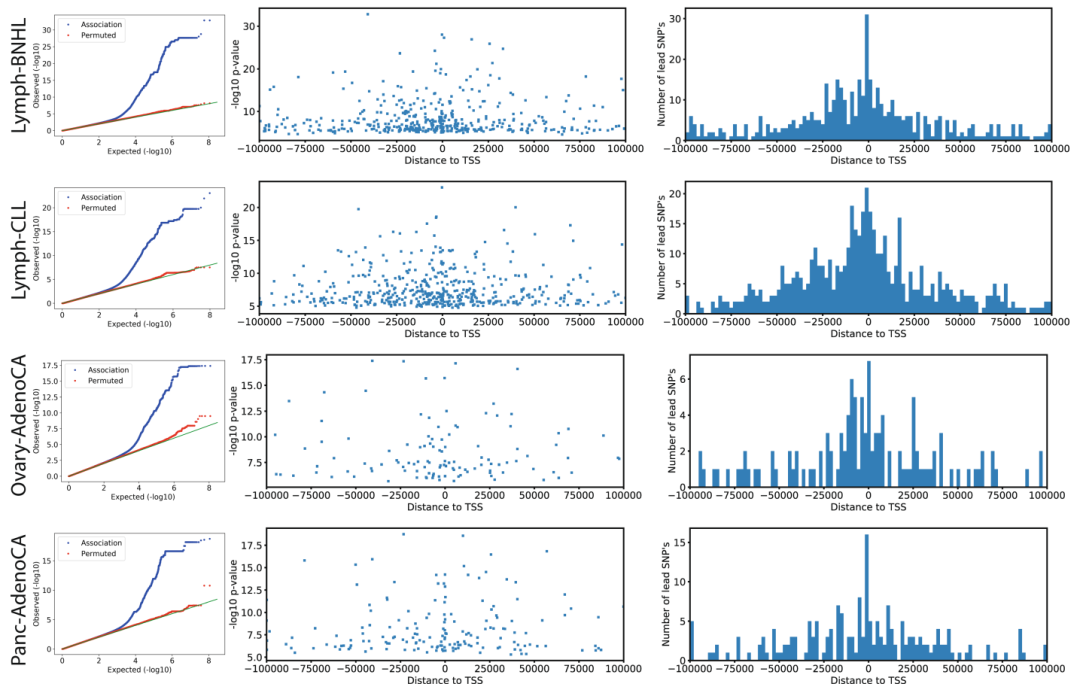
该泛癌分析发现了3,532个eQTL基因 (假发现率,即FDR≤5%,以下表示为eGenes) (Supplementary Table 2),富集于转录起始位点的近端区域 (TSSs) (Extended Data Fig. 3)。
为了识别癌症特异性调控变异,我们将我们的eQTL与来自基因型-组织表达项目 (GTEx,数据一般来自健康组织)的eQTL进行比较,采用之前的策略来评估eQTL的Replication,并探索先导eQTL变异在GTEx组织中的边缘意义 (P≤0.01, Bonferroni-adjusted)。
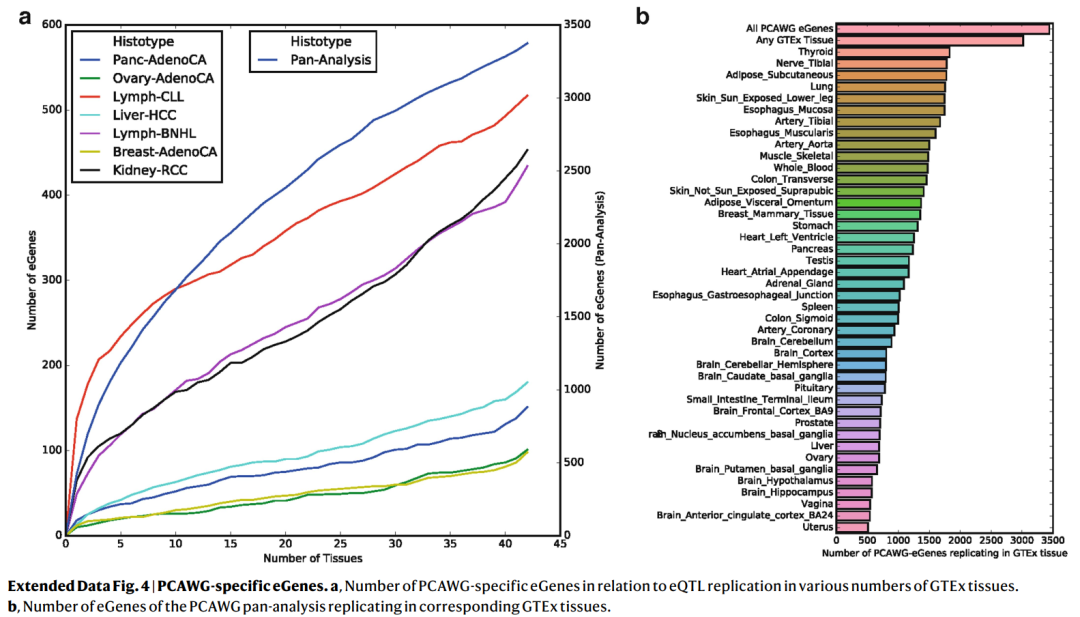
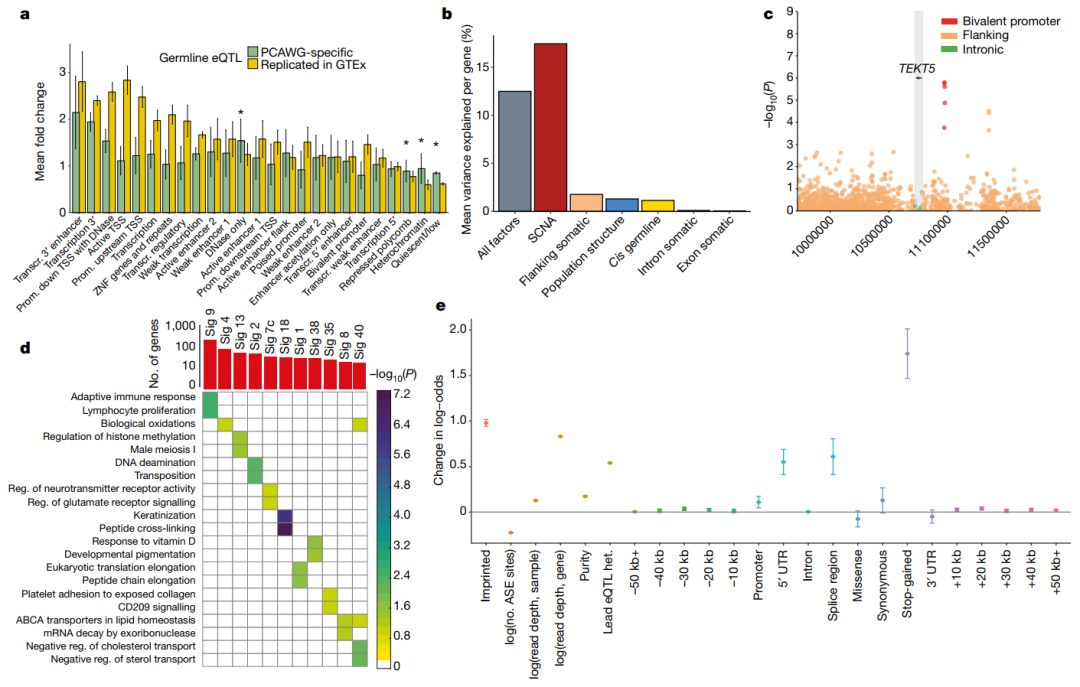
尽管大多数先导变异在GTEx样本中都能检测到 (3,532个eQTL变异中有3,110个),但我们鉴定出了422个 (~8.4%)与GTEx组织不对应的eQTL,这提示了存在癌症特异性调控 (Extended Data Fig. 4, Supplementary Table 3)。相应的eQTL先导变异富集于异染色质区 (Heterochromatic region) (图1a)。总的来说,这一分析揭示了基因表达调控的胚系框架 (Germline framework)在癌症组织中很大程度上是保守的。
Somatic cis-eQTLs in non-coding regions
先前的其它研究已经描述了癌症中的非编码突变,特别是在启动子区,及其对基因表达的调控作用。在这里,我们研究了整个基因组中,可能的体细胞DNA变化,这些变化是基因表达变化的基础。
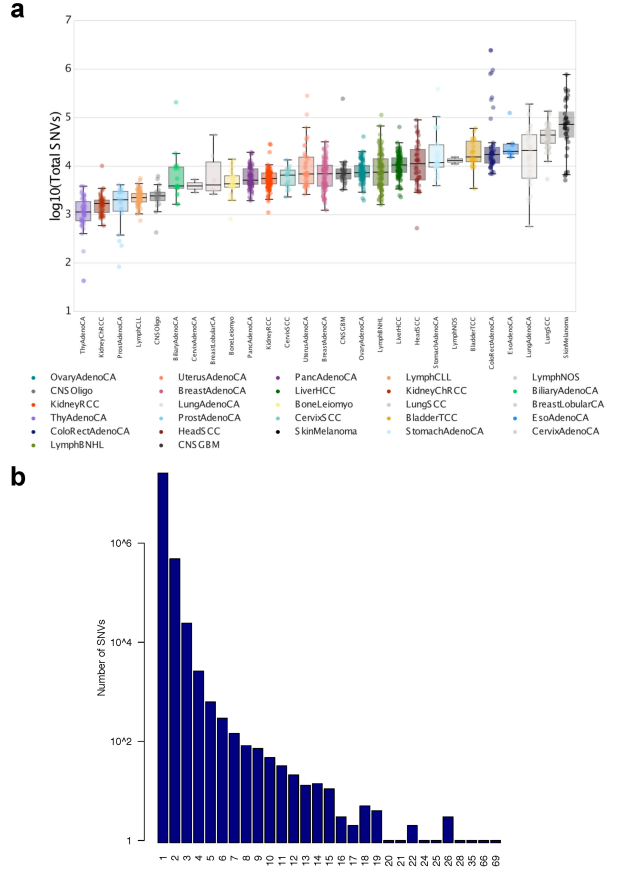
Extended Data Fig. 5 | 顺式突变体细胞负担 (Cis-mutational somatic burden)
我们通过聚集 (Aggregating)基因附近 (侧翼)2 kb区间 (2-kb intervals adjacent to genes, flanking)的SNV,以及处在外显子、内含子中的SNV (Extended Data Figs. 2, 5, 6),来估计局部突变负荷 (Estimat local mutation burden)。
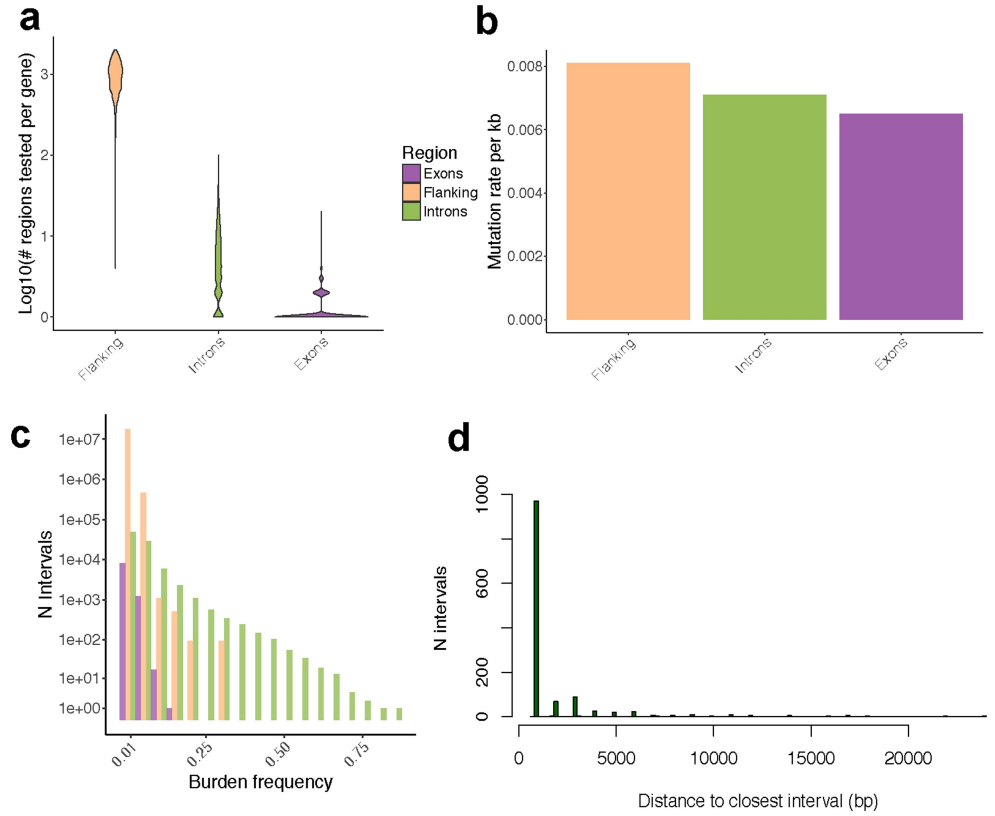
接下来,我们分解 (Decomposed)了单个基因的表达变化,考虑了顺式基因中常见的突变负荷,以及顺式胚系变异和体细胞拷贝数改变 (SCNAs)。这表明SCNAs是表达变化的主要驱动因素 (17%),其次是基因侧翼区域的体细胞SNV (1.8%)和胚系变异 (1.3%) (图1b)。
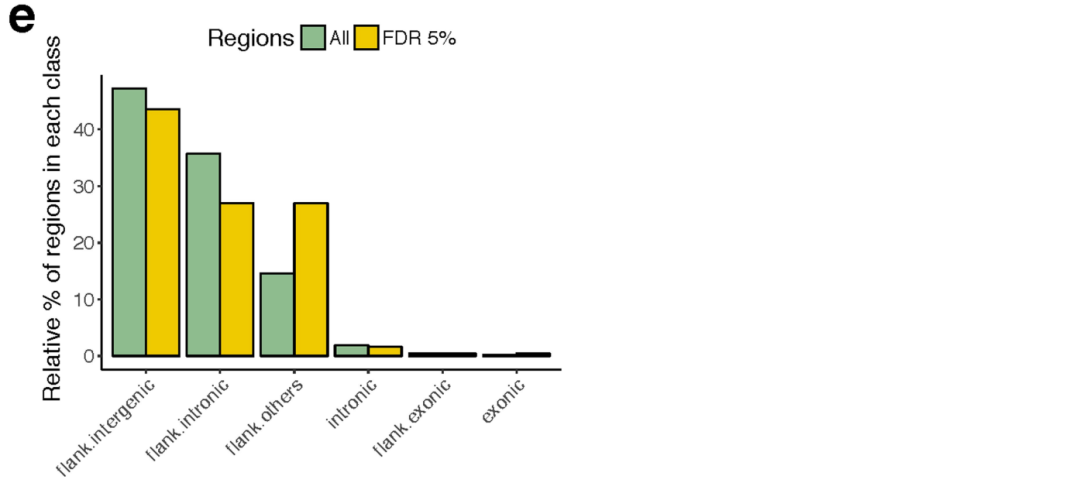
我们还测试了所有常见突变负荷和整个基因组的基因表达之间的关联。我们鉴定了649个具有体细胞eQTL (FDR≤5%)的基因 (Supplementary Table 5)。其中,11个关联结果位于相应eGene的内含子或外显子,包括在特定癌症发病机制中已知存在作用的基因,如卵巢癌中的CDK12和慢性淋巴细胞白血病中的IRF4 (Extended Data Figs. 7, 8)。
大多数eQTL (68.4%)与侧边非编码突变负荷相关 (Extended Data Fig. 6e)。
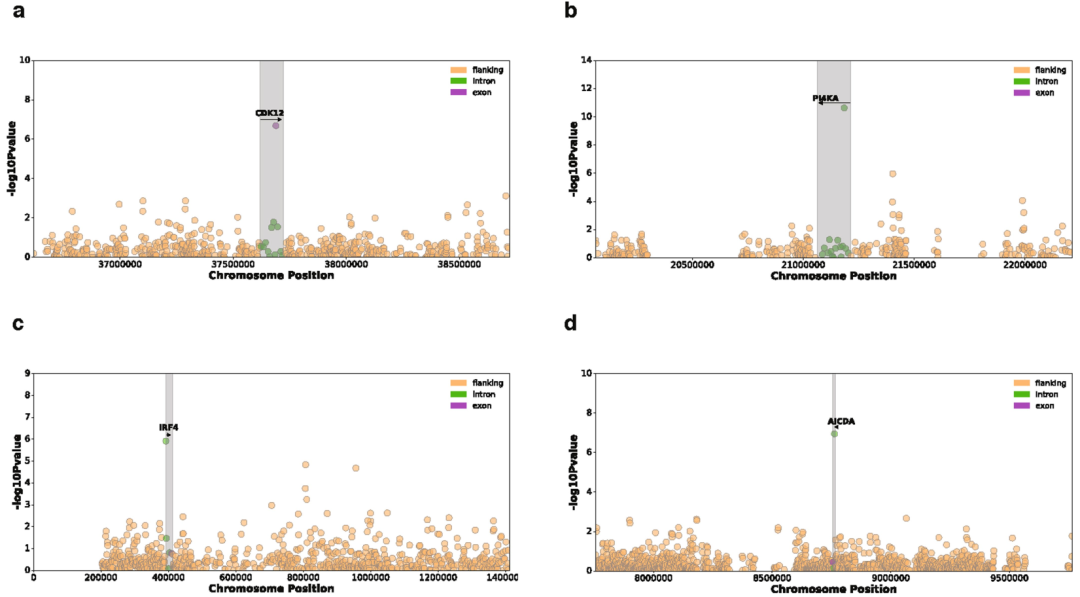
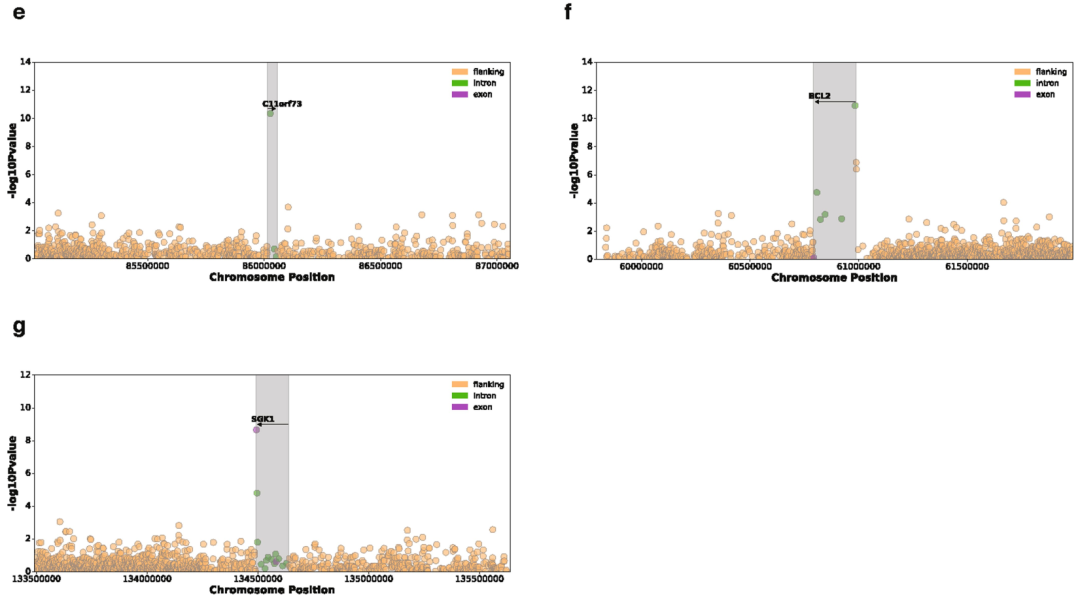
Extended Data Fig. 7 | 与遗传先导负荷 (Genic lead burden)相关联的7个体细胞eGenes的曼哈顿图
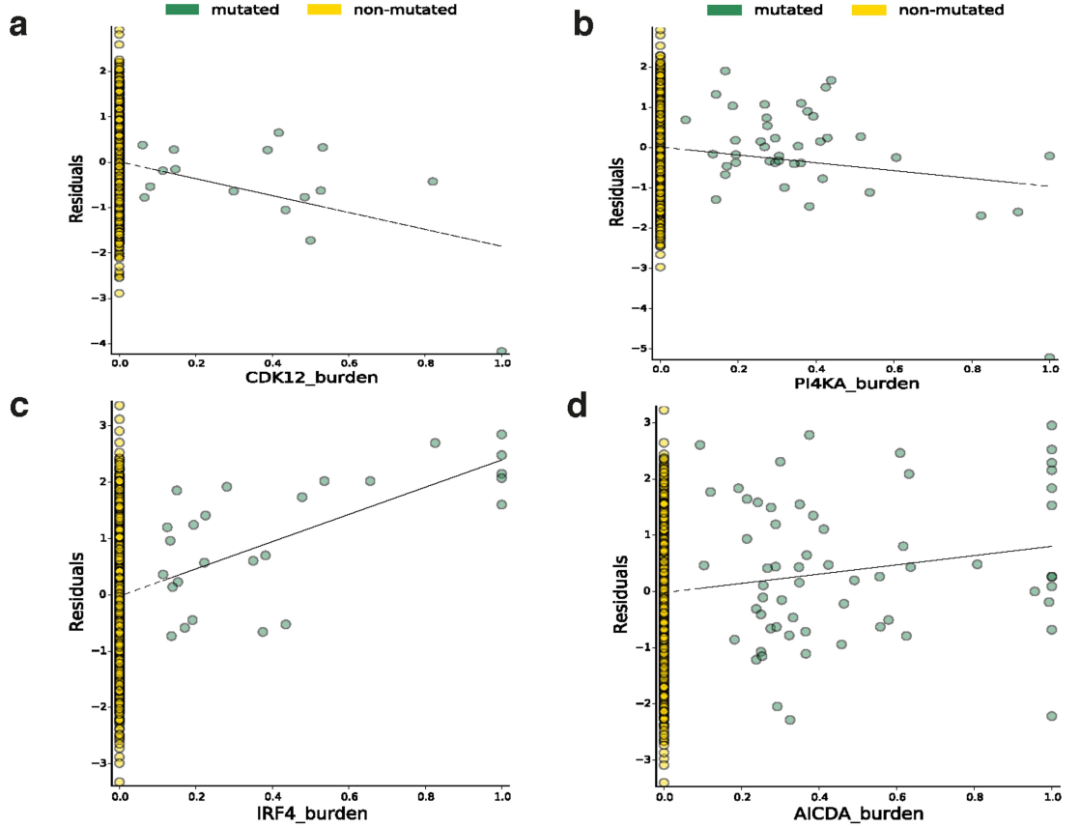
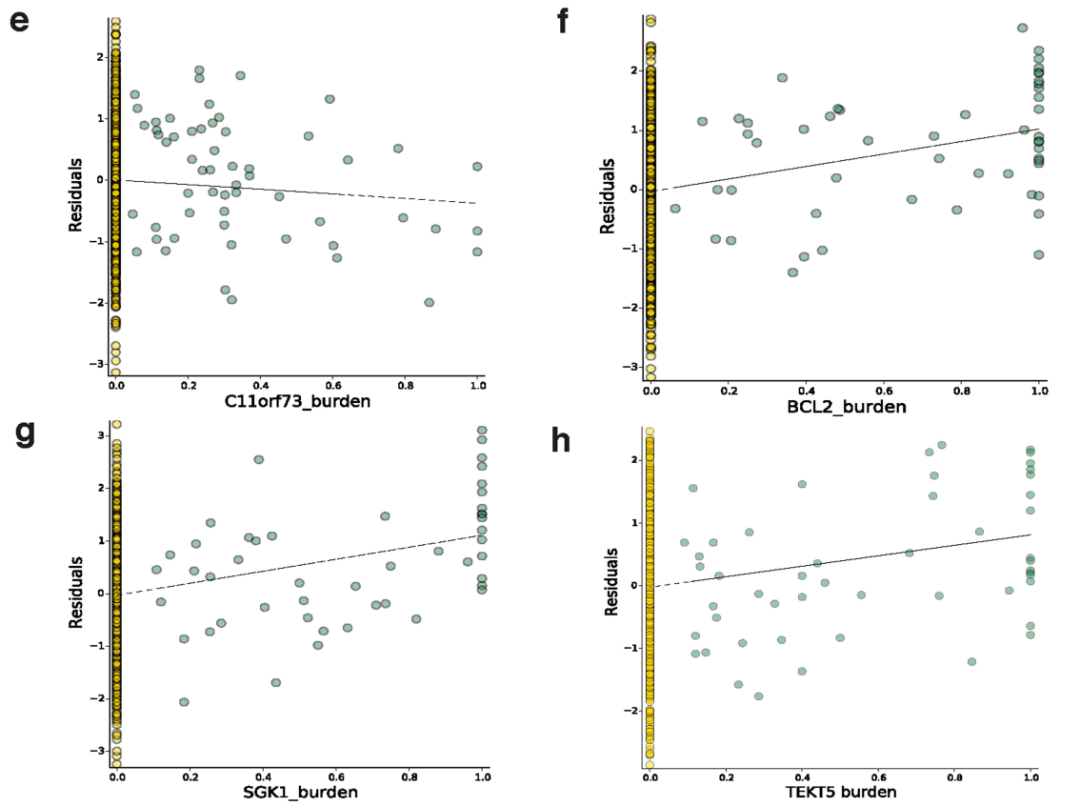
Extended Data Fig. 8 | 8个体细胞eGenes的散点图
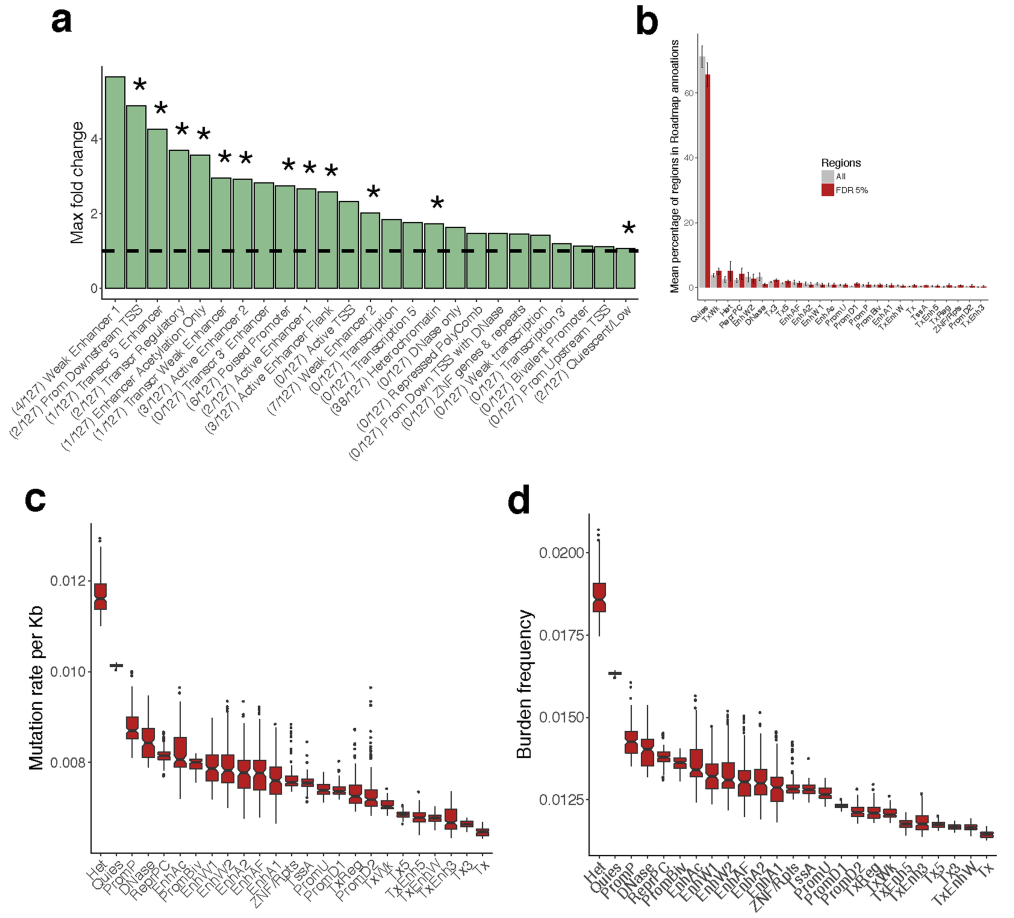
接下来,我们考虑了位于侧翼区域 (n = 556)的eQTLs,并测试了来自Epigenetics Roadmap的细胞类型特异性注释的富集。这确定了13个富集的注释突变 (FDR≤10%) (Extended Data Fig. 9, Supplementary Table 6),包括待发 (Poised)启动子,弱的和活跃的增强子,以及异染色质,但明显没有富集到转录因子结合位点 (Supplementary Table 7)。转录不活跃区域的富集可能是由于这些区域的突变率增加 (Extended Data Fig. 9),之前在癌症中有报道。
我们还研究了体细胞eGenes的功能特征,并观察到癌细胞testis基因的二价 (Bivalent)启动子中体细胞eQTLs的富集 (P = 0.04, Fisher’s exact test),如TEKT518 (Fig. 1c, Extended Data Fig. 8h)。
此外,我们发现了与细胞分化和发育过程相关的基因本体 (即GO)类别的整体 (Global)富集 (FDR≤10%) (Supplementary Table 8)。总体而言,体细胞eQTL分析发现,大多数非编码区域与局部基因表达的变化相关,与癌症特异性胚系eQTL类似,显示了转录非活性区域的富集,如异染色质。
未完 (约剩余80%的内容)
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集