SCENIC | 从单细胞数据推断基因调控网络和细胞类型
在2019/08/07的Nature刊中,中科院景乃禾课题组发表了文章——Molecular architecture of lineage allocation and tissue organization in early mouse embryo ,我在这篇文章中发现了一个被汤神组 (就是Hemberg-lab单细胞转录组数据分析(二)- 实验平台中开辟了单细胞转录组领域的人)反复用到的R工具-SCENIC,现在让我们来一起看看该工具有何妙用!
SCENIC简介
SCENIC是一种同时重建基因调控网络并从单细胞RNA-seq数据中鉴定stable cell states的工具。基于共表达和DNA模基序 (motif)分析推断基因调控网络 ,然后在每个细胞中分析网络活性以鉴定细胞状态。
SCENIC发表于2017年的Nature method文章。具体见链接:
https://www.nature.com/articles/nmeth.4463

要求:当前版本的SCENIC支持人类,鼠和果蝇(Drosophila melanogaster)。
要将SCENIC应用于其他物种,需要手动调整第二步(例如使用新的RcisTarget数据库或使用不同的motif-enrichment-analysis工具)。
输入:SCENIC需要输入的是单细胞RNA-seq表达矩阵—— 每列对应于样品(细胞),每行对应一个基因。基因ID应该是gene-symbol并存储为rownames (尤其是基因名字部分是为了与RcisTarget数据库兼容);表达数据是Gene的reads count。根据作者的测试,提供原始的或Normalized UMI count,无论是否log转换,或使用TPM值,结果相差不大。(Overall, SCENIC is quite robust to this choice, we have applied SCENIC to datasets using raw (logged) UMI counts, normalized UMI counts, and TPM and they all provided reliable results (see Aibar et al. (2017)).)
SCENIC
先安装R,如果处理大样本数据,则建议使用Python版 (https://pyscenic.readthedocs.io/en/latest/);
SCENIC在R中实现基于三个R包:
GENIE3:推断基因共表达网络
RcisTarget:用于分析转录因子结合motif
AUCell:用于鉴定scRNA-seq数据中具有活性基因集(基因网络)的细胞
运行SCENIC需要安装这些软件包以及一些额外的依赖包:
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::version()
# If your bioconductor version is previous to 3.9, see the section bellow
## Required
BiocManager::install(c("AUCell", "RcisTarget"))
BiocManager::install(c("GENIE3")) # Optional. Can be replaced by GRNBoost
## Optional (but highly recommended):
# To score the network on cells (i.e. run AUCell):
BiocManager::install(c("zoo", "mixtools", "rbokeh"))
# For various visualizations and perform t-SNEs:
BiocManager::install(c("DT", "NMF", "pheatmap", "R2HTML", "Rtsne"))
# To support paralell execution (not available in Windows):
BiocManager::install(c("doMC", "doRNG"))
# To export/visualize in http://scope.aertslab.org
if (!requireNamespace("devtools", quietly = TRUE)) install.packages("devtools")
devtools::install_github("aertslab/SCopeLoomR", build_vignettes = TRUE)
Version: AUCell >=1.4.1 (minimum 1.2.4), RcisTarget>=1.2.0 (minimum 1.0.2) and GENIE3>=1.4.0(minimum 1.2.1).
安装SCENIC:
if (!requireNamespace("devtools", quietly = TRUE)) install.packages("devtools")
devtools::install_github("aertslab/SCENIC")
packageVersion("SCENIC")下载评分数据库
除了必要的R包之外,需要下载RcisTarget的物种特定数据库(https://resources.aertslab.org/cistarget/;主题排名)。默认情况下,SCENIC使用在基因启动子(TSS上游500 bp)和TSS周围 20 kb (+/- 10kb)中对模序进行评分的数据库。
For human:
dbFiles <- c("https://resources.aertslab.org/cistarget/databases/homo_sapiens/hg19/refseq_r45/mc9nr/gene_based/hg19-500bp-upstream-7species.mc9nr.feather",
"https://resources.aertslab.org/cistarget/databases/homo_sapiens/hg19/refseq_r45/mc9nr/gene_based/hg19-tss-centered-10kb-7species.mc9nr.feather")
# mc9nr: Motif collection version 9: 24k motifsFor mouse:
dbFiles <- c("https://resources.aertslab.org/cistarget/databases/mus_musculus/mm9/refseq_r45/mc9nr/gene_based/mm9-500bp-upstream-7species.mc9nr.feather",
"https://resources.aertslab.org/cistarget/databases/mus_musculus/mm9/refseq_r45/mc9nr/gene_based/mm9-tss-centered-10kb-7species.mc9nr.feather")
# mc9nr: Motif collection version 9: 24k motifs
For fly:
dbFiles <- c("https://resources.aertslab.org/cistarget/databases/drosophila_melanogaster/dm6/flybase_r6.02/mc8nr/gene_based/dm6-5kb-upstream-full-tx-11species.mc8nr.feather")
# mc8nr: Motif collection version 8: 20k motifs
注意:下载后最好确认下载的数据是否完整,可基于MD5值评估。(参考链接:https://resources.aertslab.org/cistarget/databases/sha256sum.txt)
完成这些设置步骤后,SCENIC即可运行。
数据格式不同时如何读入?
最终读入的信息有两个,一个是前面说的表达矩阵,还有一个是样品分组信息。
a) From
.loomfile
.loom文件可以通过SCopeLoomR包直接导入SCENIC。(loom格式是用于存储非常大的组学数据集的专属格式,具体见 http://linnarssonlab.org/loompy/)
## Download:
download.file("http://loom.linnarssonlab.org/clone/Previously%20Published/Cortex.loom", "Cortex.loom")
loomPath <- "Cortex.loom"b) From 10X/CellRanger output files
10X/CellRanger输出结果可用作SCENIC的输入文件 (需要先安装Seurat)。
# BiocManager::install("Seurat")
# 测试数据也可以从Seurat官网下载,自己修改路径
# 测试数据:https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/output/matrices#r-load-mat>
singleCellMatrix <- Seurat::Read10X(data.dir="data/pbmc3k/filtered_gene_bc_matrices/hg19/")
cellInfo <- data.frame(seuratCluster=Idents(seuratObject))c) From other R objects (e.g. Seurat, SingleCellExperiment)
sce <- load_as_sce(dataPath) # any SingleCellExperiment object
exprMat <- counts(sce)
cellInfo <- colData(sce)d) From GEO
从GEO下载和格式化数据集的示例(例如,GEOGSE60361:3005小鼠脑细胞数据集)
# 获取数据及数据标注
# dir.create("SCENIC_MouseBrain"); setwd("SCENIC_MouseBrain") # if needed
# (This may take a few minutes)
if (!requireNamespace("GEOquery", quietly = TRUE)) BiocManager::install("GEOquery")
library(GEOquery)
geoFile <- getGEOSuppFiles("GSE60361", makeDirectory=FALSE)
gzFile <- grep("Expression", basename(rownames(geoFile)), value=TRUE)
txtFile <- gsub(".gz", "", gzFile)
gunzip(gzFile, destname=txtFile, remove=TRUE)
library(data.table)
geoData <- fread(txtFile, sep="\t")
geneNames <- unname(unlist(geoData[,1, with=FALSE]))
exprMatrix <- as.matrix(geoData[,-1, with=FALSE])
rm(geoData)
dim(exprMatrix)
rownames(exprMatrix) <- geneNames
exprMatrix <- exprMatrix[unique(rownames(exprMatrix)),]
exprMatrix[1:5,1:4]
# Remove file downloaded:
file.remove(txtFile)
运行 SCENIC
SCENIC workflow:
建立基因调控网络(Gene Regulation Network,GRN):
基于共表达识别每个转录因子TF的潜在靶标。
过滤表达矩阵并运行GENIE3或者GRNBoost,它们是利用表达矩阵推断基因调控网络的一种算法,能得到转录因子和潜在靶标的相关性网络;
将目标从GENIE3或者GRNBoost格式转为共表达模块。根据DNA模序分析(
motif)选择潜在的直接结合靶标(调节因子)(利用RcisTarget包:TF基序分析)
确定细胞状态及其调节因子:
3. 分析每个细胞中的网络活性(AUCell)
在细胞中评分调节子(计算AUC)
SCENIC完整流程
### 导入数据
loomPath<-system.file(package="SCENIC","examples/mouseBrain_toy.loom")
library(SCopeLoomR)
loom <- open_loom(loomPath, mode="r")
exprMat <- get_dgem(loom)
cellInfo <- get_cellAnnotation(loom)
close_loom(loom)
### Initialize settings 初始设置,导入评分数据库
library(SCENIC)
scenicOptions <- initializeScenic(org="mgi", dbDir="cisTarget_databases", nCores=10)
# scenicOptions@inputDatasetInfo$cellInfo <- "int/cellInfo.Rds"
saveRDS(scenicOptions, file="int/scenicOptions.Rds")
### 共表达网络
genesKept <- geneFiltering(exprMat, scenicOptions)
exprMat_filtered <- exprMat[genesKept, ]
runCorrelation(exprMat_filtered, scenicOptions)
exprMat_filtered_log <- log2(exprMat_filtered+1)
runGenie3(exprMat_filtered_log, scenicOptions)
### Build and score the GRN 构建并给基因调控网络(GRN)打分
exprMat_log <- log2(exprMat+1)
scenicOptions@settings$dbs <- scenicOptions@settings$dbs["10kb"] # Toy run settings
runSCENIC_1_coexNetwork2modules(scenicOptions)
runSCENIC_2_createRegulons(scenicOptions, coexMethod=c("top5perTarget")) # Toy run settings
runSCENIC_3_scoreCells(scenicOptions, exprMat_log)
# Export:
export2scope(scenicOptions, exprMat)
# Binarize activity?
# aucellApp <- plotTsne_AUCellApp(scenicOptions, exprMat_log)
# savedSelections <- shiny::runApp(aucellApp)
# newThresholds <- savedSelections$thresholds
# scenicOptions@fileNames$int["aucell_thresholds",1] <- "int/newThresholds.Rds"
# saveRDS(newThresholds, file=getIntName(scenicOptions, "aucell_thresholds"))
# saveRDS(scenicOptions, file="int/scenicOptions.Rds")
runSCENIC_4_aucell_binarize(scenicOptions)
### Exploring output
# Check files in folder 'output'
# .loom file @ http://scope.aertslab.org
# output/Step2_MotifEnrichment_preview.html in detail/subset:
motifEnrichment_selfMotifs_wGenes <- loadInt(scenicOptions, "motifEnrichment_selfMotifs_wGenes")
tableSubset <- motifEnrichment_selfMotifs_wGenes[highlightedTFs=="Sox8"]
viewMotifs(tableSubset)
# output/Step2_regulonTargetsInfo.tsv in detail:
regulonTargetsInfo <- loadInt(scenicOptions, "regulonTargetsInfo")
tableSubset <- regulonTargetsInfo[TF=="Stat6" & highConfAnnot==TRUE]
viewMotifs(tableSubset)
举个栗子!
输入表达矩阵
在本教程中,我们提供了一个示例,样本是小鼠大脑的200个细胞和862个基因:
loomPath <- system.file(package="SCENIC", "examples/mouseBrain_toy.loom")打开loom文件并加载表达矩阵;
library(SCopeLoomR)
loom <- open_loom(loomPath, mode="r")
exprMat <- get_dgem(loom)
cellInfo <- get_cellAnnotation(loom)
close_loom(loom)
dim(exprMat)
细胞信息/表型
# cellInfo$nGene <- colSums(exprMat>0)
head(cellInfo)
cellInfo <- data.frame(cellInfo)
cellTypeColumn <- "Class"
colnames(cellInfo)[which(colnames(cellInfo)==cellTypeColumn)] <- "CellType"
cbind(table(cellInfo$CellType))
# Color to assign to the variables (same format as for NMF::aheatmap)
colVars <- list(CellType=c("microglia"="forestgreen",
"endothelial-mural"="darkorange",
"astrocytes_ependymal"="magenta4",
"oligodendrocytes"="hotpink",
"interneurons"="red3",
"pyramidal CA1"="skyblue",
"pyramidal SS"="darkblue"))
colVars$CellType <- colVars$CellType[intersect(names(colVars$CellType), cellInfo$CellType)]
saveRDS(colVars, file="int/colVars.Rds")
plot.new(); legend(0,1, fill=colVars$CellType, legend=names(colVars$CellType))
初始化SCENIC设置
为了在SCENIC的多个步骤中保持设置一致,SCENIC包中的大多数函数使用一个公共对象,该对象存储当前运行的选项并代替大多数函数的“参数”。比如下面的org,dbDir等,可以在开始就将物种rog(mgi—— mouse, hgnc —— human, dmel —— fly)和RcisTarge数据库位置分别读给对象org,dbDir,之后统一用函数initializeScenic得到对象scenicOptions。具体参数设置可以用?initializeScenichelp一下。
library(SCENIC)
org="mgi" # or hgnc, or dmel
dbDir="cisTarget_databases" # RcisTarget databases location
myDatasetTitle="SCENIC example on Mouse brain" # choose a name for your analysis
data(defaultDbNames)
dbs <- defaultDbNames[[org]]
scenicOptions <- initializeScenic(org=org, dbDir=dbDir, dbs=dbs, datasetTitle=myDatasetTitle, nCores=10)
# Modify if needed
scenicOptions@inputDatasetInfo$cellInfo <- "int/cellInfo.Rds"
scenicOptions@inputDatasetInfo$colVars <- "int/colVars.Rds"
# Databases:
# scenicOptions@settings$dbs <- c("mm9-5kb-mc8nr"="mm9-tss-centered-5kb-10species.mc8nr.feather")
# scenicOptions@settings$db_mcVersion <- "v8"
# Save to use at a later time...
saveRDS(scenicOptions, file="int/scenicOptions.Rds")共表达网络
SCENIC工作流程的第一步是根据表达数据推断潜在的转录因子靶标。为此,我们使用GENIE3或GRNBoost,输入文件是表达矩阵(过滤后的)和转录因子列表。GENIE3/GRBBoost的输出结果和相关矩阵将用于创建共表达模块(runSCENIC_1_coexNetwork2modules())。
基因过滤/选择
按每个基因的reads总数进行过滤。
该filter旨在去除最可能是噪音的基因。
默认情况下,它(
minCountsPerGene)保留所有样品中至少带有6个UMI reads的基因(例如,如果在1%的细胞中以3的值表达,则基因将具有的总数)。通过基因的细胞数来实现过滤(例如
UMI > 0,或log 2(TPM)> 1 )。默认情况下(
minSamples),保留下来的基因能在至少1%的细胞中检测得到。最后,只保留
RcisTarget数据库中可用的基因。
# (Adjust minimum values according to your dataset)
genesKept <- geneFiltering(exprMat, scenicOptions=scenicOptions,
minCountsPerGene=3*.01*ncol(exprMat),
minSamples=ncol(exprMat)*.01)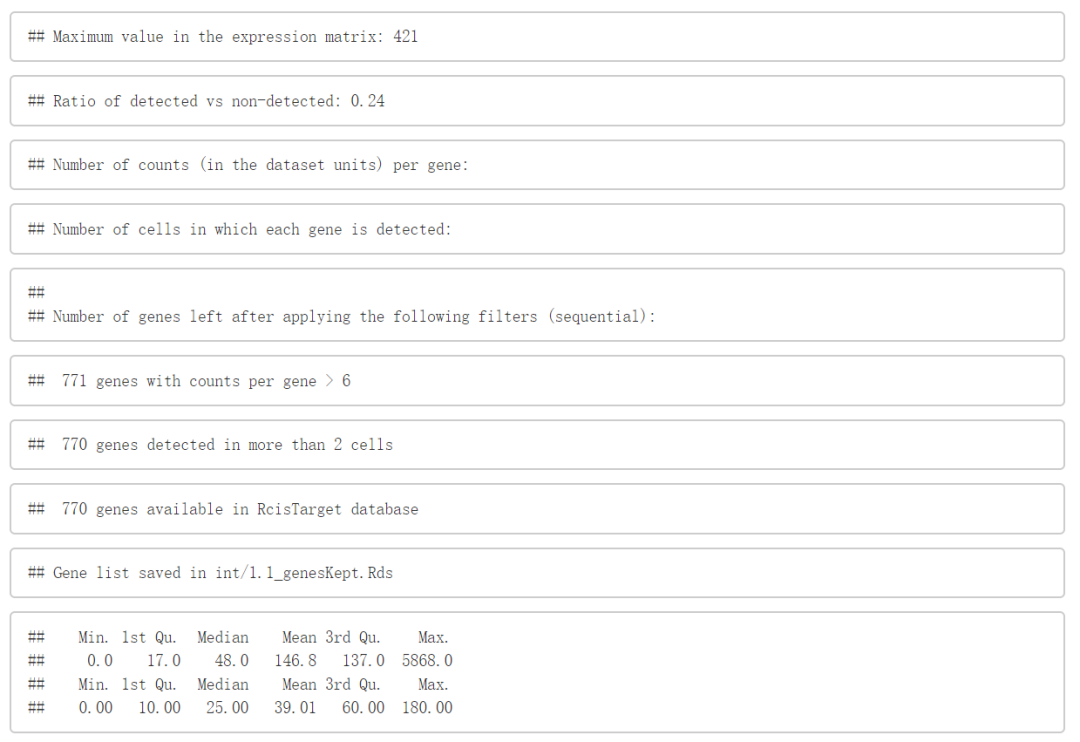
在进行网络推断之前,检查是否有任何已知的相关基因被过滤掉(如果缺少任何相关基因,请仔细检查filter设置是否合适):
interestingGenes <- c("Sox9", "Sox10", "Dlx5")
# any missing?
interestingGenes[which(!interestingGenes %in% genesKept)]
相关性
GENIE33或者GRNBoost可以检测正负关联。为了区分潜在的激活和抑制,我们将目标分为正相关和负相关目标(比如TF与潜在目标之间的Spearman相关性)。
runCorrelation(exprMat_filtered, scenicOptions)运行GENIE3得到潜在转录因子TF
## If launched in a new session, you will need to reload...
# setwd("...")
# loomPath <- "..."
# loom <- open_loom(loomPath, mode="r")
# exprMat <- get_dgem(loom)
# close_loom(loom)
# genesKept <- loadInt(scenicOptions, "genesKept")
# exprMat_filtered <- exprMat[genesKept,]
# library(SCENIC)
# scenicOptions <- readRDS("int/scenicOptions.Rds")
# Optional: add log (if it is not logged/normalized already)
exprMat_filtered <- log2(exprMat_filtered+1)
# Run GENIE3
runGenie3(exprMat_filtered, scenicOptions)
构建并评分GRN(runSCENIC_ …)
必要时重新加载表达式矩阵:
loom <- open_loom(loomPath, mode="r")
exprMat <- get_dgem(loom)
close_loom(loom)
# Optional: log expression (for TF expression plot, it does not affect any other calculation)
logMat <- log2(exprMat+1)
dim(exprMat)
使用wrapper函数运行其余步骤:
library(SCENIC)
scenicOptions <- readRDS("int/scenicOptions.Rds")
scenicOptions@settings$verbose <- TRUE
scenicOptions@settings$nCores <- 10
scenicOptions@settings$seed <- 123
# For a very quick run:
# coexMethod=c("top5perTarget")
scenicOptions@settings$dbs <- scenicOptions@settings$dbs["10kb"] # For toy run
# save...
runSCENIC_1_coexNetwork2modules(scenicOptions)
runSCENIC_2_createRegulons(scenicOptions, coexMethod=c("top5perTarget")) #** Only for toy run!!
runSCENIC_3_scoreCells(scenicOptions, logMat)
可选步骤
将network activity转换成ON/OFF(二进制)格式
nPcs <- c(5) # For toy dataset
# nPcs <- c(5,15,50)scenicOptions@settings$seed <- 123 # same seed for all of them
# Run t-SNE with different settings:
fileNames <- tsneAUC(scenicOptions, aucType="AUC", nPcs=nPcs, perpl=c(5,15,50))
fileNames <- tsneAUC(scenicOptions, aucType="AUC", nPcs=nPcs, perpl=c(5,15,50), onlyHighConf=TRUE, filePrefix="int/tSNE_oHC")
# Plot as pdf (individual files in int/):
fileNames <- paste0("int/",grep(".Rds", grep("tSNE_", list.files("int"), value=T), value=T))
par(mfrow=c(length(nPcs), 3))
fileNames <- paste0("int/",grep(".Rds", grep("tSNE_AUC", list.files("int"), value=T, perl = T), value=T))
plotTsne_compareSettings(fileNames, scenicOptions, showLegend=FALSE, varName="CellType", cex=.5)

# Using only "high-confidence" regulons (normally similar)
par(mfrow=c(3,3))
fileNames <- paste0("int/",grep(".Rds", grep("tSNE_oHC_AUC", list.files("int"), value=T, perl = T), value=T))
plotTsne_compareSettings(fileNames, scenicOptions, showLegend=FALSE, varName="CellType", cex=.5)
输出到 loom/SCope
SCENIC生成的结果既能在http://scope.aertslab.org查看,还能用函数export2scope()(需要SCopeLoomR包)保存成.loom文件。
# DGEM (Digital gene expression matrix)
# (non-normalized counts)
# exprMat <- get_dgem(open_loom(loomPath))
# dgem <- exprMat
# head(colnames(dgem)) #should contain the Cell ID/name
# Export:
scenicOptions@fileNames$output["loomFile",] <- "output/mouseBrain_SCENIC.loom"
export2scope(scenicOptions, exprMat)
加载.loom文件中的结果
SCopeLoomR中也有函数可以导入.loom文件中的内容,比如调节因子,AUC和封装内容(比如regulon activity的t-SNE和UMAP结果)。
library(SCopeLoomR)
scenicLoomPath <- getOutName(scenicOptions, "loomFile")
loom <- open_loom(scenicLoomPath)
# Read information from loom file:
regulons_incidMat <- get_regulons(loom)
regulons <- regulonsToGeneLists(regulons_incidMat)
regulonsAUC <- get_regulonsAuc(loom)
regulonsAucThresholds <- get_regulonThresholds(loom)
embeddings <- get_embeddings(loom)
解读结果
1. 细胞状态
AUCell提供跨细胞的调节子的活性,AUCell使用“Area under Curve 曲线下面积”(AUC)来计算输入基因集的关键子集是否在每个细胞的表达基因中富集。通过该调节子活性(连续或二进制AUC矩阵)来聚类细胞,我们可以看出是否存在倾向于具有相同调节子活性的细胞群,并揭示在多个细胞中反复发生的网络状态。这些状态等同于网络的吸引子状态。将这些聚类与不同的可视化方法相结合,我们可以探索细胞状态与特定调节子的关联。
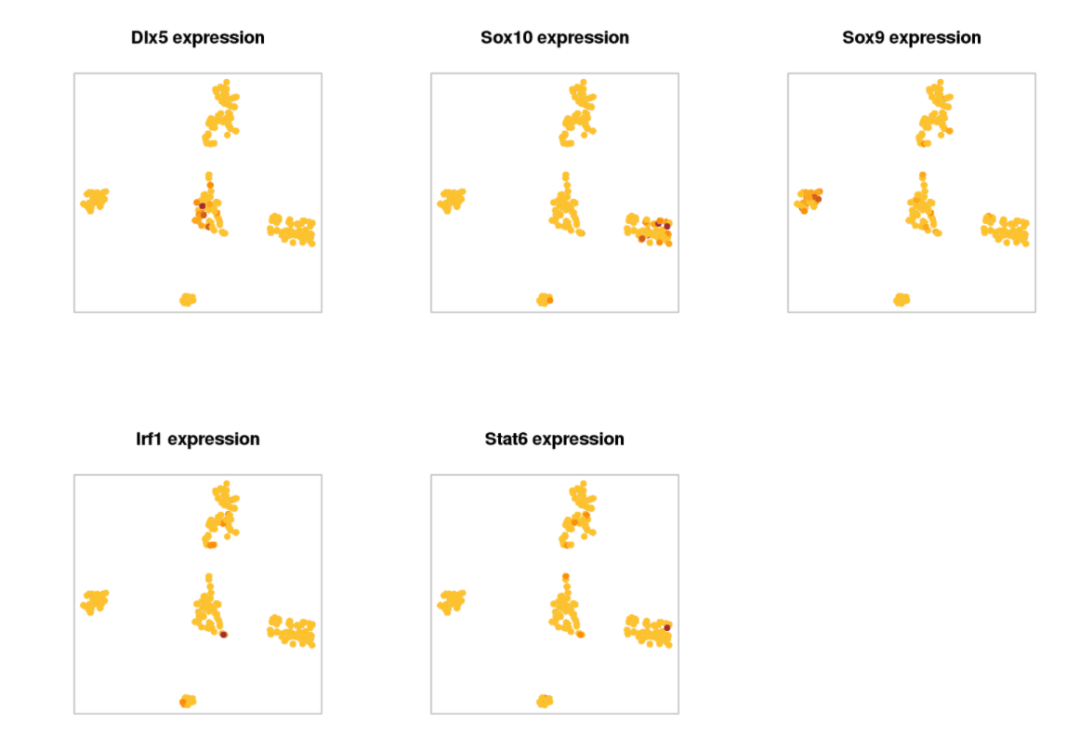
将AUC和TF表达投射到t-SNE上
logMat <- exprMat # Better if it is logged/normalized
aucellApp <- plotTsne_AUCellApp(scenicOptions, logMat) # default t-SNE
savedSelections <- shiny::runApp(aucellApp)
print(tsneFileName(scenicOptions))tSNE_scenic <- readRDS(tsneFileName(scenicOptions))
aucell_regulonAUC <- loadInt(scenicOptions, "aucell_regulonAUC")
# Show TF expression:
par(mfrow=c(2,3))
AUCell::AUCell_plotTSNE(tSNE_scenic$Y, exprMat, aucell_regulonAUC[onlyNonDuplicatedExtended(rownames(aucell_regulonAUC))[c("Dlx5", "Sox10", "Sox9","Irf1", "Stat6")],], plots="Expression")
# Save AUC as PDF:
Cairo::CairoPDF("output/Step4_BinaryRegulonActivity_tSNE_colByAUC.pdf", width=20, height=15)
par(mfrow=c(4,6))
AUCell::AUCell_plotTSNE(tSNE_scenic$Y, cellsAUC=aucell_regulonAUC, plots="AUC")
dev.off()
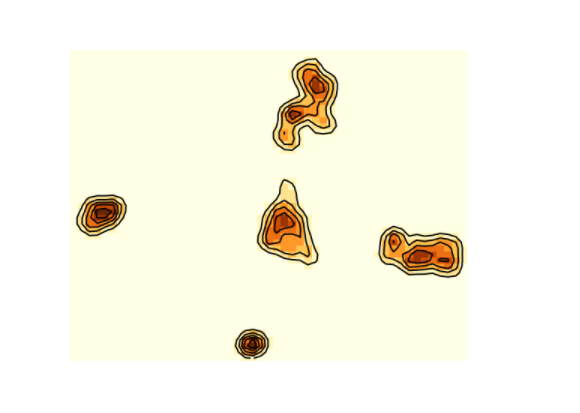
library(KernSmooth)library(RColorBrewer)
dens2d <- bkde2D(tSNE_scenic$Y, 1)$fhat
image(dens2d, col=brewer.pal(9, "YlOrBr"), axes=FALSE)
contour(dens2d, add=TRUE, nlevels=5, drawlabels=FALSE)
#par(bg = "black")
par(mfrow=c(1,2))
regulonNames <- c( "Dlx5","Sox10")
cellCol <- plotTsne_rgb(scenicOptions, regulonNames, aucType="AUC", aucMaxContrast=0.6)
text(0, 10, attr(cellCol,"red"), col="red", cex=.7, pos=4)
text(-20,-10, attr(cellCol,"green"), col="green3", cex=.7, pos=4)
regulonNames <- list(red=c("Sox10", "Sox8"),
green=c("Irf1"),
blue=c( "Tef"))
cellCol <- plotTsne_rgb(scenicOptions, regulonNames, aucType="Binary")
text(5, 15, attr(cellCol,"red"), col="red", cex=.7, pos=4)
text(5, 15-4, attr(cellCol,"green"), col="green3", cex=.7, pos=4)
text(5, 15-8, attr(cellCol,"blue"), col="blue", cex=.7, pos=4)
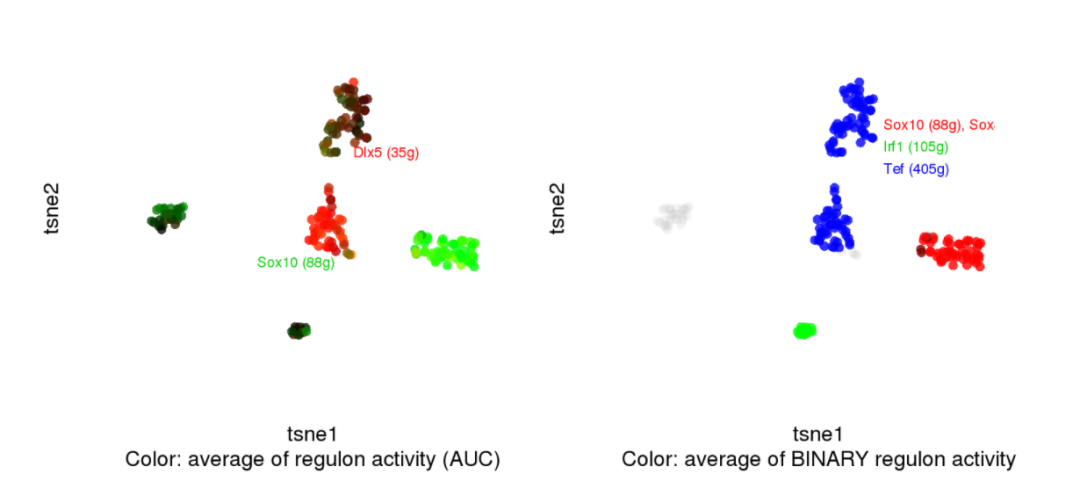
GRN:Regulon靶标和模序
regulons <- loadInt(scenicOptions, "regulons")
regulons[c("Dlx5", "Irf1")]

regulons <- loadInt(scenicOptions, "aucell_regulons")
head(cbind(onlyNonDuplicatedExtended(names(regulons))))

regulonTargetsInfo <- loadInt(scenicOptions, "regulonTargetsInfo")
tableSubset <- regulonTargetsInfo[TF=="Stat6" & highConfAnnot==TRUE]
viewMotifs(tableSubset)
2. 细胞群的调控因子
regulonAUC <- loadInt(scenicOptions, "aucell_regulonAUC")
regulonAUC <- regulonAUC[onlyNonDuplicatedExtended(rownames(regulonAUC)),]
regulonActivity_byCellType <- sapply(split(rownames(cellInfo), cellInfo$CellType),
function(cells) rowMeans(getAUC(regulonAUC)[,cells]))
regulonActivity_byCellType_Scaled <- t(scale(t(regulonActivity_byCellType), center = T, scale=T))
pheatmap::pheatmap(regulonActivity_byCellType_Scaled, #fontsize_row=3,
color=colorRampPalette(c("blue","white","red"))(100), breaks=seq(-3, 3, length.out = 100),
treeheight_row=10, treeheight_col=10, border_color=NA)
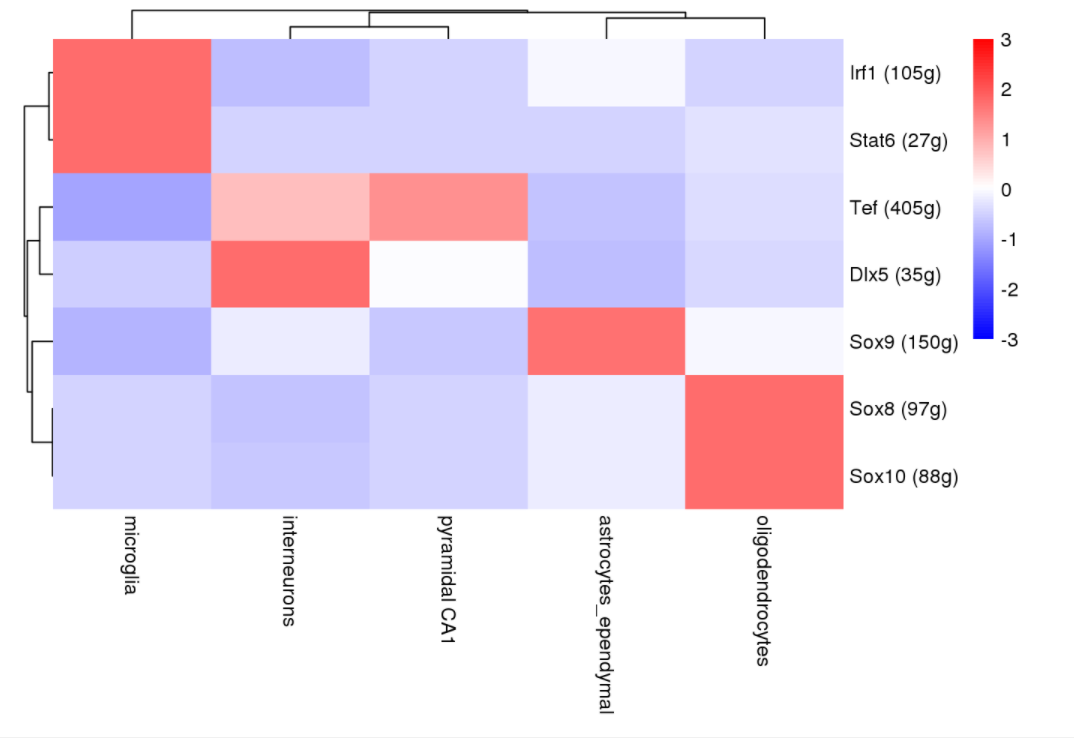
# filename="regulonActivity_byCellType.pdf", width=10, height=20)
topRegulators <- reshape2::melt(regulonActivity_byCellType_Scaled)
colnames(topRegulators) <- c("Regulon", "CellType", "RelativeActivity")
topRegulators <- topRegulators[which(topRegulators$RelativeActivity>0),]
viewTable(topRegulators)
minPerc <- .7
binaryRegulonActivity <- loadInt(scenicOptions, "aucell_binary_nonDupl")
cellInfo_binarizedCells <- cellInfo[which(rownames(cellInfo)%in% colnames(binaryRegulonActivity)),, drop=FALSE]
regulonActivity_byCellType_Binarized <- sapply(split(rownames(cellInfo_binarizedCells), cellInfo_binarizedCells$CellType),
function(cells) rowMeans(binaryRegulonActivity[,cells, drop=FALSE]))
binaryActPerc_subset <- regulonActivity_byCellType_Binarized[which(rowSums(regulonActivity_byCellType_Binarized>minPerc)>0),]
pheatmap::pheatmap(binaryActPerc_subset, # fontsize_row=5,
color = colorRampPalette(c("white","pink","red"))(100), breaks=seq(0, 1, length.out = 100),
treeheight_row=10, treeheight_col=10, border_color=NA)
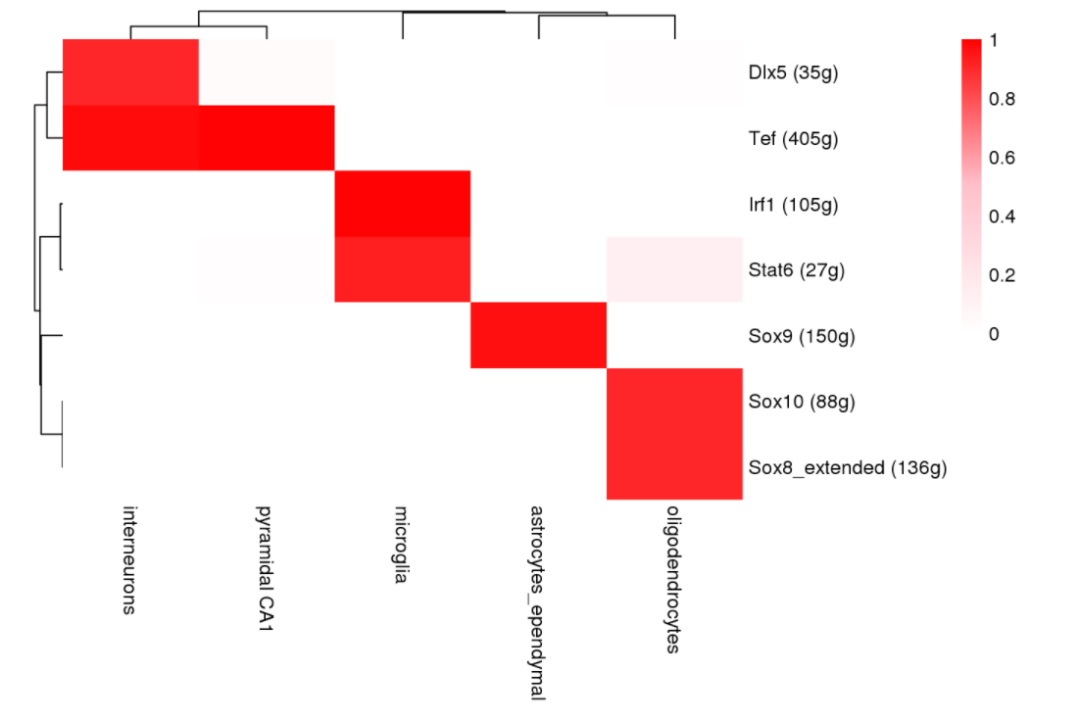
参考文献
Aibar, Sara, Carmen Bravo González-Blas, Thomas Moerman, Jasper Wouters, Vân Anh Huynh-Thu, Hana Imrichová, Zeynep Kalender Atak, et al. 2017. “SCENIC: Single-Cell Regulatory Network Inference and Clustering.” Nature Methods 14 (october): 1083–6. doi:10.1038/nmeth.4463.
Davie, Kristofer, Jasper Janssens, Duygu Koldere, and “et al.” 2018. “A Single-Cell Transcriptome Atlas of the Aging Drosophila Brain.” Cell, june. doi:10.1016/j.cell.2018.05.057.
Huynh-Thu, Vân Anh, Alexandre Irrthum, Louis Wehenkel, and Pierre Geurts. 2010. “Inferring Regulatory Networks from Expression Data Using Tree-Based Methods.” PloS One 5 (9). doi:10.1371/journal.pone.0012776.
Marbach, Daniel, James C. Costello, Robert Küffner, Nicole M. Vega, Robert J. Prill, Diogo M. Camacho, Kyle R. Allison, et al. 2012. “Wisdom of Crowds for Robust Gene Network Inference.” Nature Methods 9 (8): 796–804. doi:10.1038/nmeth.2016.
https://rawcdn.githack.com/aertslab/SCENIC/0a4c96ed8d930edd8868f07428090f9dae264705/inst/doc/SCENIC_Running.html#directories
你可能还想看
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集