本文约3200字,建议阅读9分钟
本文介绍了清华团队在单细胞技术的最新进展。
单细胞技术的最新进展使得能够在细胞水平上表征表观基因组异质性。鉴于细胞数量呈指数增长,迫切需要用于自动细胞类型注释的计算方法。特别是,单细胞染色质可及性测序(scCAS)数据的注释,它可以捕获控制每种细胞类型转录的染色质调控景观;然而,科学家对该领域的研究却少之又少。
清华大学的研究人员提出了 EpiAnno,一种与贝叶斯神经网络集成的概率生成模型,以有监督的方式自动注释 scCAS 数据。他们系统地验证了 EpiAnno 在各种数据集中,展现出的数据集内和数据集间注释的卓越性能。
通过表达富集分析、分区遗传力分析、增强子识别、顺式可访问性分析和通路富集分析,进一步证明了 EpiAnno 在可解释嵌入和生物学意义方面的优势。此外,研究人员表明 EpiAnno 具有揭示细胞类型特异性基序并促进 scCAS 数据模拟的潜力。
该研究以「Cell type annotation of single-cell chromatin accessibility data via supervised Bayesian embedding」为题,于 2022 年 2 月 10 日发布在《Nature Machine Intelligence》。

背景
单细胞测序技术的最新创新为揭示以前未知的细胞类型异质性提供了前所未有的机会,并彻底改变了对各种复杂组织的理解。为了识别单细胞数据集中存在的细胞群,典型的方法是无监督细胞聚类,然后将推定的细胞类型标签分配给每个聚类。
然而,随着被分析的单个细胞呈指数级增长,由于依赖于手动注释,这种方法变得繁琐、不可复制且耗时,手动注释通常不是基于细胞标签的标准本体,而是高度依赖于研究者关于特征分子的背景知识。因此,需要对单个细胞进行自动细胞类型注释。
目前,学界已经提出了几种专门用于单细胞 RNA 测序(scRNA-seq)数据的细胞类型注释的计算方法。这些方法可以分为三大类:
- 基于相似性的方法,例如scmap,它根据每个细胞与参考细胞之间的相似性将细胞映射到预定义的细胞类型;
- 基于标记基因的方法,例如 Garnett,通过以标记基因的形式结合先验知识来注释细胞类型;
- 基于监督学习的方法,例如 SuperCT,它通过机器学习方法和带注释的参考 scRNA-seq 数据对细胞进行分类。
尽管如此,绝大多数工作都集中在单细胞基因表达上,但尚未对单细胞染色质可及性测序(scCAS)数据的注释进行充分研究。染色质可及性作为核大分子物理接触 DNA 能力的量度,对于理解调控机制至关重要。scCAS 数据的细胞类型注释可以捕获控制每种细胞类型转录的染色质调控环境。然而,scCAS 数据注释比 scRNA-seq 数据注释更具挑战性,因为(1)文献中的指南较少,(2)分析DNA时的低拷贝数导致仅捕获1-10%的潜在可访问区域,(3)与scRNA-seq数据中的基因不同,scCAS数据中捕获的可访问区域因数据集而异,(4)scCAS数据具有特定于分析的挑战,包括其接近二进制性质、极端稀疏性以及比scRNA-seq数据高几十倍的维度。因此,scCAS数据注释通常通过三个主要步骤进行:- 通过聚集每个基因转录起始位点(TSS)周围的可访问性得分,计算scCAS数据的基因水平染色质可访问性得分;
- 使用scRNA-seq特异性方法通过细胞基质对获得的基因的细胞类型标签进行注释;
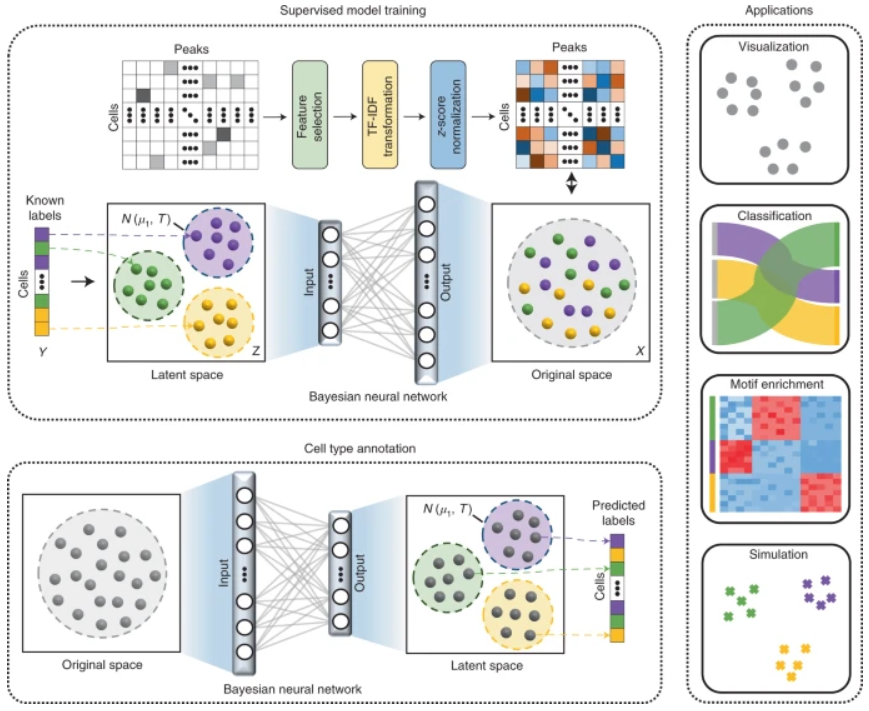
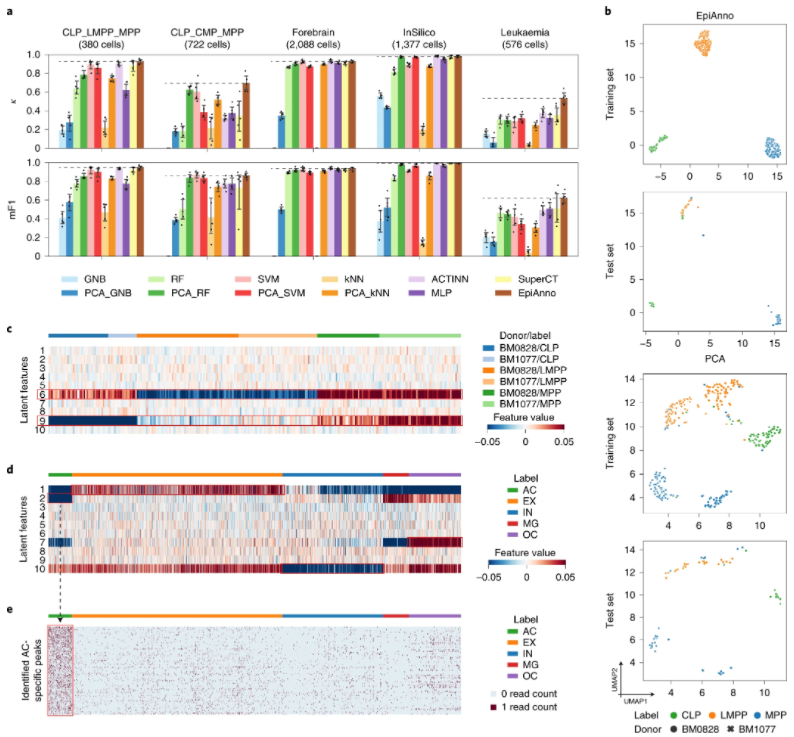
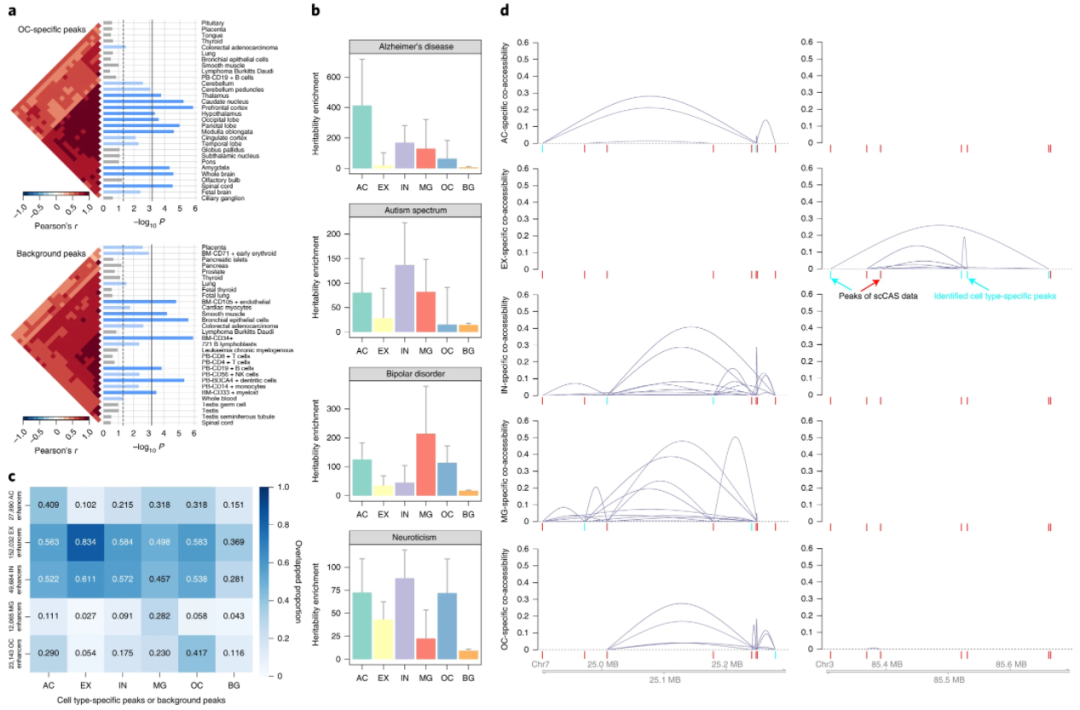
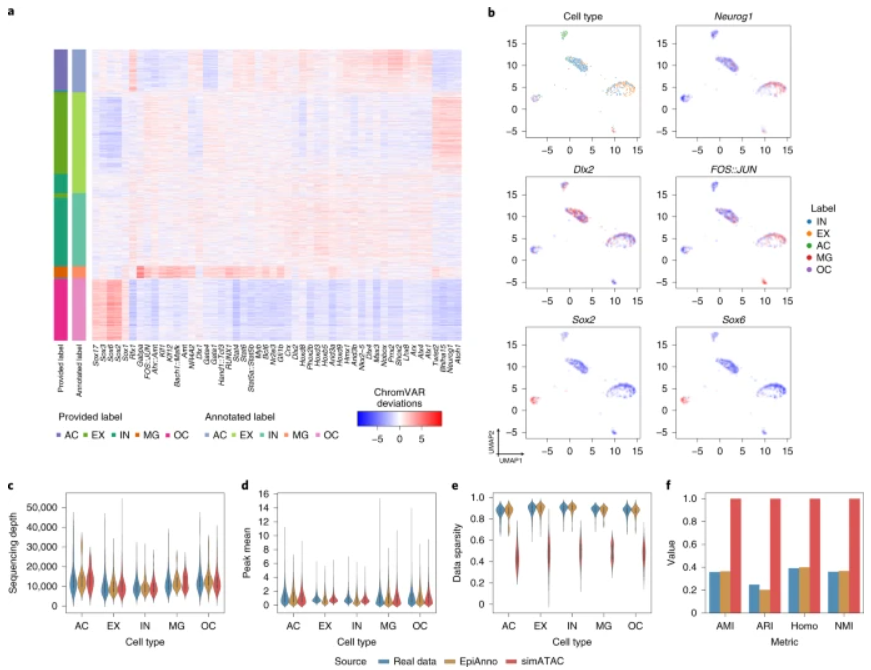
直观地说,由于可访问性分数的聚合,上述方法均不能充分利用 scCAS 数据中的固有信息,这表明对 scCAS 数据的自动细胞类型注释的迫切需求。在该研究中,研究人员提出了概率生成模型 EpiAnno,以更好地表征细胞异质性并准确注释 scCAS 数据中的细胞类型。EpiAnno 使用贝叶斯神经网络将细胞嵌入到潜在空间中,其中细胞遵循高斯混合分布(每种细胞类型对应于潜在高斯分布)。EpiAnno 是一种基于模型的方法,用于在 scCAS 数据中表征细胞并以监督方式注释细胞类型。为了降低 scCAS 数据的噪声水平,研究人员首先采用了类似于最近研究的峰值选择策略。然后,在实施模型之前,研究人员执行了词频-逆文档频率(TF-IDF)转换,并计算了 z 分数以标准化 scCAS 数据矩阵。注意,数据处理是在训练和测试集中独立进行的。如图 1 所示,从概率生成的角度说明了:EpiAnno 首先根据给定的单元类型标签,从一个潜在高斯分布中导出每个单元的潜在表示,然后使用非线性贝叶斯神经网络将嵌入投影到scCAS数据的原始特征/峰值空间。贝叶斯神经网络表征高维 scCAS 数据并提供嵌入数据和归一化数据之间的可解释关联,而潜在高斯混合分布用作基于细胞嵌入对细胞进行分类的判别模型。潜在高斯分布的数量等于参考/训练数据集中的细胞类型数量,并且 EpiAnno 的默认潜在维度设置为 10,如 SCALE 中一样。借助变分推理算法和集成学习策略,EpiAnno 可高效稳定地执行参数估计。图 1:EpiAnno 通过有监督的贝叶斯嵌入对 scCAS 数据的细胞类型进行注释。(来源:论文)研究人员首先使用数据集内的注释作为概念证明,来展示 EpiAnno 的性能。他们收集了人类造血细胞数据集(称为 CLP_LMPP_MPP 数据集),进行了五折交叉验证实验,将所有细胞随机分成五折,并使用剩余四折训练的模型迭代预测每一折中细胞的细胞类型标签。使用 Cohen 的 kappa 值 (κ) 和中值 F1 分数 (mF1) 来评估注释性能。EpiAnno 以11种基线方法为基准;其结果如图 2a 所示,EpiAnno 实现了最佳且稳定的标注性能,而基线方法的性能在不同数据集之间波动。为了进一步证明 EpiAnno 在可解释注释方面的优势,研究人员对小鼠前脑的转座酶可访问染色质 (ATAC)-seq 数据集进行了单核分析。EpiAnno获得了令人满意的注释性能,并捕获了具有特定潜在特征的不同细胞类型。然后,研究人员根据相应特征(方法)的负载确定了特定于细胞类型的峰,并可视化了每个细胞在这些峰中的读取计数。- 首先,EpiAnno 鉴定的细胞类型特异性峰提供了组织特异性表达富集。
- 第三,EpiAnno 可以帮助定义候选细胞类型特异性调控元件,从而为基因调控机制提供新的见解。
- 第四,EpiAnno 有助于揭示特定细胞类型的共同可访问站点。
- 第五,EpiAnno 识别的细胞类型特异性峰可以揭示细胞亚群的功能含义。
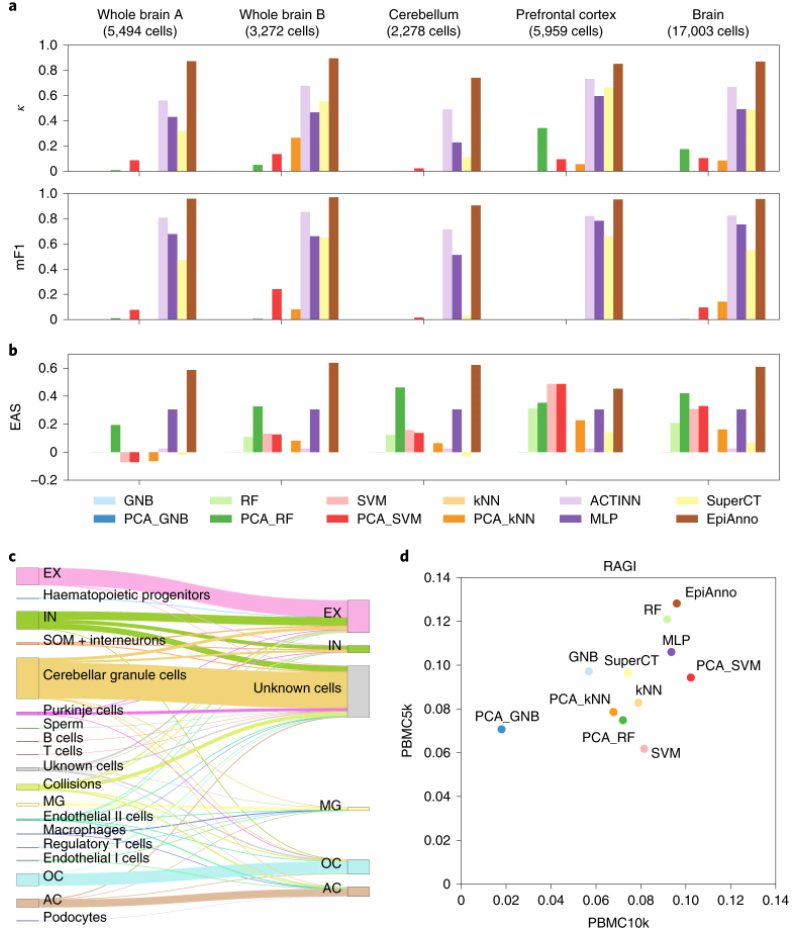
图 3:EpiAnno 确定的细胞类型特异性峰的生物学意义。(来源:论文)总而言之,EpiAnno 不仅可以准确注释 scCAS 数据中的细胞,还可以提供可解释的细胞嵌入和生物学意义。然后,研究人员将 EpiAnno 应用于数据集间标注,这是一个更实际的应用场景。与大多数特征/基因在数据集之间共享的 scRNA-seq 数据不同,scCAS 数据的特征/峰值因数据集而异。为了对 scCAS 数据启用有监督的数据集间注释,研究人员统一了训练集和测试集的峰值,并关注训练集和测试集之间共享的细胞类型的注释性能,以及共享单元类型和测试集合唯一单元类型的注释性能。如图 4 所示,EpiAnno 在这两个数据集上取得了整体最佳性能,进一步表明 EpiAnno 可以在更实际的数据集间注释场景中有效地注释细胞类型。基序富集分析是阐明特定环境的调节机制的基础。研究人员使用全脑A数据集的细胞来说明 EpiAnno(数据集间注释)的注释结果可以有效地揭示细胞类型特定的基序。研究人员对 EpiAnno 注释的共享细胞类型进行了基序富集,并可视化了前 50 个可变转录因子(TF)结合基序。丰富的 TF 结合基序提供了直观的细胞类型特异性可视化模式;进一步证实了这些 TF/基序在相应细胞类型中的作用。图 5 :基序富集分析和 scCAS 数据模拟。(来源:论文)已经为 scCAS 数据分析开发了一系列计算方法和流程,需要合成和可重复的数据集来指导实验设计和基准测试稳健性。作为一种生成方法,经过训练的 EpiAnno 模型可用于在与训练数据相同的生物学条件下生成高保真合成数据。在上述数据集间实验中,研究人员使用前脑数据集训练的 EpiAnno 模型,为每种类型的细胞生成峰值矩阵,其细胞数量与训练数据中的细胞数相同。使用相同的训练数据,研究人员还通过最先进的 scCAS 数据模拟框架 simATAC 模拟了每种类型的相同数量的细胞。与 simATAC 类似,研究人员通过测序深度、数据稀疏性、峰值均值和细胞聚类来评估了模拟性能。基于对多个数据集的综合实验,研究人员证明 EpiAnno 在 scCAS 数据的数据集内和跨数据集注释方面均优于基线方法。与大多数计算方法不同,经过训练的 EpiAnno 参数和学习的细胞嵌入是可解释的,并且可以通过组织特异性表达富集分析、分区遗传力分析、细胞类型特异性增强子识别和细胞类型特异性 cis-coaccessibility 分析揭示生物学见解。此外,研究人员发现 EpiAnno 不仅具有揭示细胞类型特异性基序的潜力,而且为 scCAS 数据的模拟开辟了一条新途径,这对于 scCAS 数据分析方法的发展至关重要。论文链接:
https://www.nature.com/articles/s42256-021-00432-w