
哈佛大学单细胞课程|笔记汇总 (八)
哈佛大学刘小乐教授讲授的计算生物学和生物信息学导论 (2020 视频+资料)
(八)Single-cell RNA-seq clustering analysis
得到高质量的细胞后,我们可以探究细胞群中的不同细胞类型。
Exploration of quality control metrics
为了确定我们的细胞亚群是否可能是由于诸如细胞周期阶段或线粒体表达之类的假象引起的,可以对不同cluster之间进行多种可视化分析判断影响因素。这里我们将调用DimPlot()和FeaturePlot()函数来分析并可视化多种质控指标。
按照样本对细胞亚群进行分离
从探索每个样品中不同细胞亚群开始分析:
# 从Seurat对象中提取身份和样本信息,以确定每个样本的每个簇的细胞数
n_cells <- FetchData(seurat_integrated,
vars = c("ident", "orig.ident")) %>%
dplyr::count(ident, orig.ident) %>%
tidyr::spread(ident, n)
# View table
View(n_cells)
使用UMAP对每个样品不同细胞亚群进行可视化:
# UMAP of cells in each cluster by sample
DimPlot(seurat_integrated,
label = TRUE,
split.by = "sample") + NoLegend()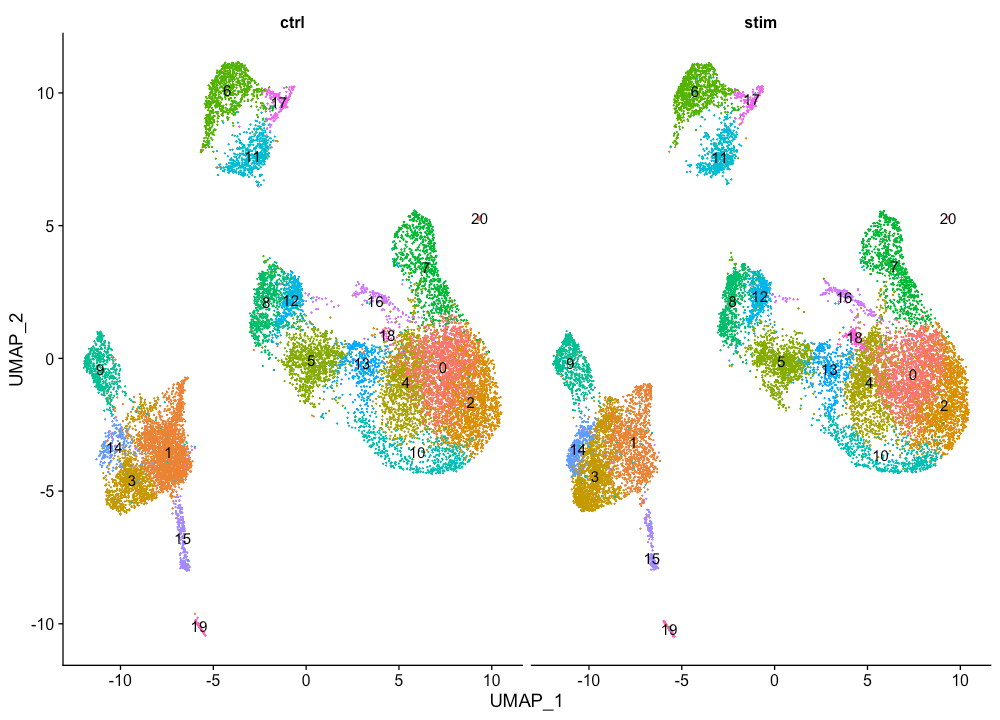
从上图没有看到单独由样品来源引起的聚类簇。
按照细胞周期对细胞亚群进行分离
我们可以探讨细胞是否在不同的细胞周期阶段聚类。当我们利用SCTransform执行归一化和无意义的变异源回归(regression of uninteresting sources of variation)时,没有考虑细胞周期的影响。
如果我们的细胞簇在不同细胞周期上显示出很大差异,则表明我们要重新运行SCTransform并将S.Score和G2M.Score (细胞周期得分)添加到变量中以进行回归,然后重新运行其余步骤 。
# Explore whether clusters segregate by cell cycle phase
DimPlot(seurat_integrated,
label = TRUE,
split.by = "Phase") + NoLegend()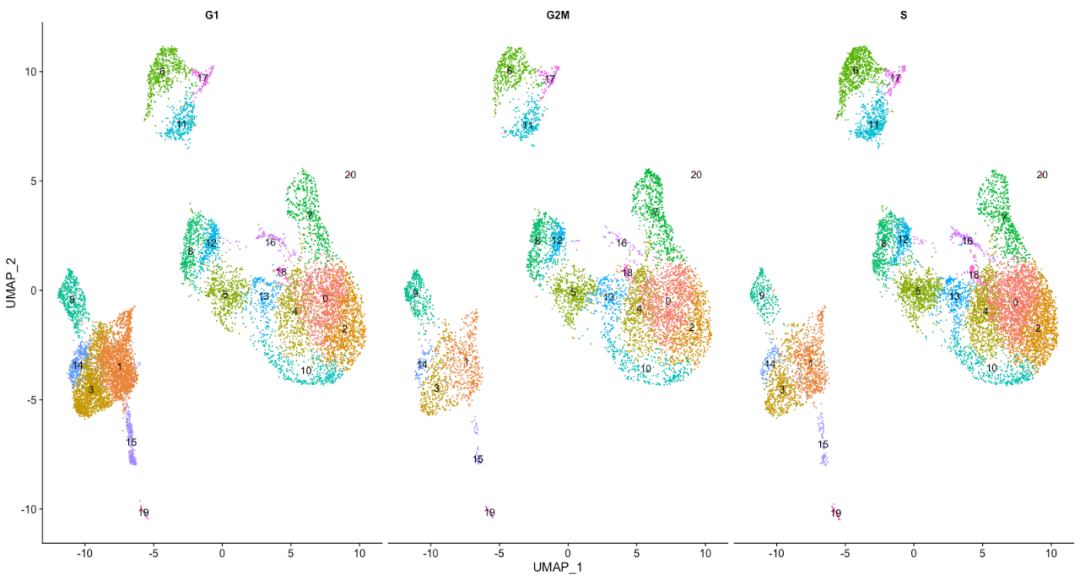
从上图未能看到单个细胞周期主导的聚类簇。
可视化其它指标对细胞亚群分群的影响
我们还可以探索其他指标,例如每个细胞的UMI和基因数量,S期和G2M期的标记,以及通过UMAP进行的线粒体基因表达。其中各个S和G2M分数可提供更多的周期信息。
# Determine metrics to plot present in seurat_integrated@meta.data
metrics <- c("nUMI", "nGene", "S.Score", "G2M.Score", "mitoRatio")
FeaturePlot(seurat_integrated,
reduction = "umap",
features = metrics,
pt.size = 0.4,
sort.cell = TRUE,
min.cutoff = 'q10',
label = TRUE)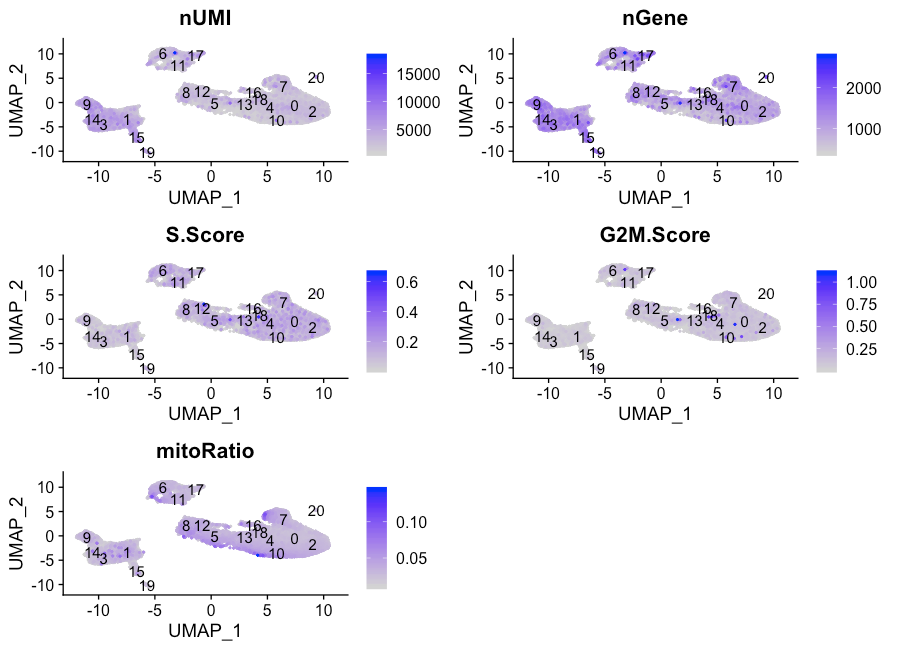
NOTE: The
sort.cellargument will plot the positive cells above the negative cells, while themin.cutoffargument will determine the threshold for shading. Amin.cutoffofq10translates to the 10% of cells with the lowest expression of the gene will not exhibit any purple shading (completely gray).
这些指标在不同的cluster中还是比较均匀的,但nUMI和nGene在cluster 3、9、14和15以及也许还有cluster 17中显示出了更高的值。
探索导致不同分群clusters的PC
我们可以探索不同PC分群clusters的情况,以确定定义的PC是否能够很好地区分细胞类型。为了可视化此信息,我们需要提取细胞的UMAP坐标信息以及每个PC的相应分数,并通过UMAP查看。
首先识别Seurat对象中的待提取信息,然后使用FetchData()函数提取。
# Defining the information in the seurat object of interest
columns <- c(paste0("PC_", 1:16),
"ident",
"UMAP_1", "UMAP_2")
# Extracting this data from the seurat object
pc_data <- FetchData(seurat_integrated,
vars = columns)NOTE: How did we know in the
FetchData()function to includeUMAP_1to obtain the UMAP coordinates? The Seurat cheatsheet(https://satijalab.org/seurat/essential_commands.html) describes the function as being able to pull any data from the expression matrices, cell embeddings, or metadata.For instance, if you explore the
seurat_integrated@reductionslist object, the first component is for PCA, and includes a slot forcell.embeddings. We can use the column names (PC_1, PC_2, PC_3, etc.) to pull out the coordinates or PC scores corresponding to each cell for each of the PCs.We could do the same thing for UMAP:
# Extract the UMAP coordinates for the first 10 cells
seurat_integrated@reductions$umap@cell.embeddings[1:10, 1:2]
The FetchData() function just allows us to extract the data more easily.利用FetchData()函数能更轻松地提取数据。
在以下UMAP图中,细胞将根据PC score进行上色。下面的是top16 PCs:
# Adding cluster label to center of cluster on UMAP
umap_label <- FetchData(seurat_integrated,
vars = c("ident", "UMAP_1", "UMAP_2")) %>%
group_by(ident) %>%
summarise(x=mean(UMAP_1), y=mean(UMAP_2))
# Plotting a UMAP plot for each of the PCs
map(paste0("PC_", 1:16), function(pc){
ggplot(pc_data,
aes(UMAP_1, UMAP_2)) +
geom_point(aes_string(color=pc),
alpha = 0.7) +
scale_color_gradient(guide = FALSE,
low = "grey90",
high = "blue") +
geom_text(data=umap_label,
aes(label=ident, x, y)) +
ggtitle(pc)
}) %>%
plot_grid(plotlist = .)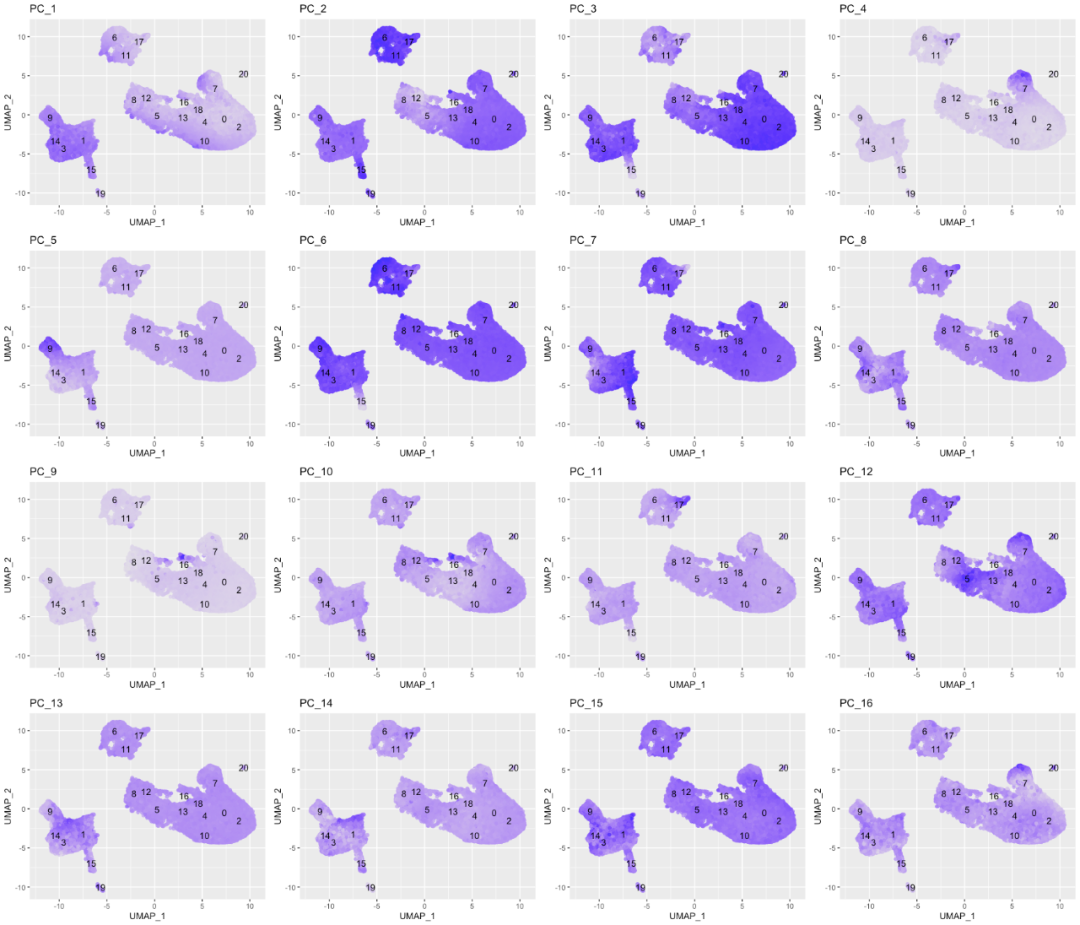
我们可以看到clusters是如何由不同的PC表示。例如,驱动PC_2的基因在cluster 6、11和17中贡献更大(在15中也可能更高),因此我们可以回顾一下驱动此PC的基因,以了解细胞类型可能是什么:
# Examine PCA results
print(seurat_integrated[["pca"]], dims = 1:5, nfeatures = 5)
使用CD79A基因和HLA基因作为PC_2的阳性标记,我们可以假设cluster 6、11和17对应于B细胞。这一点只能为我们细胞分群提供线索,cluster的真正身份还需要通过PC组合来确定。
探究已知的细胞类型marker
细胞聚类之后,我们可以通过寻找已知的marker来探索细胞类型。下图显示了带有marker的簇的UMAP图,然后显示了预期的不同细胞类型。
DimPlot(object = seurat_integrated,
reduction = "umap",
label = TRUE) + NoLegend()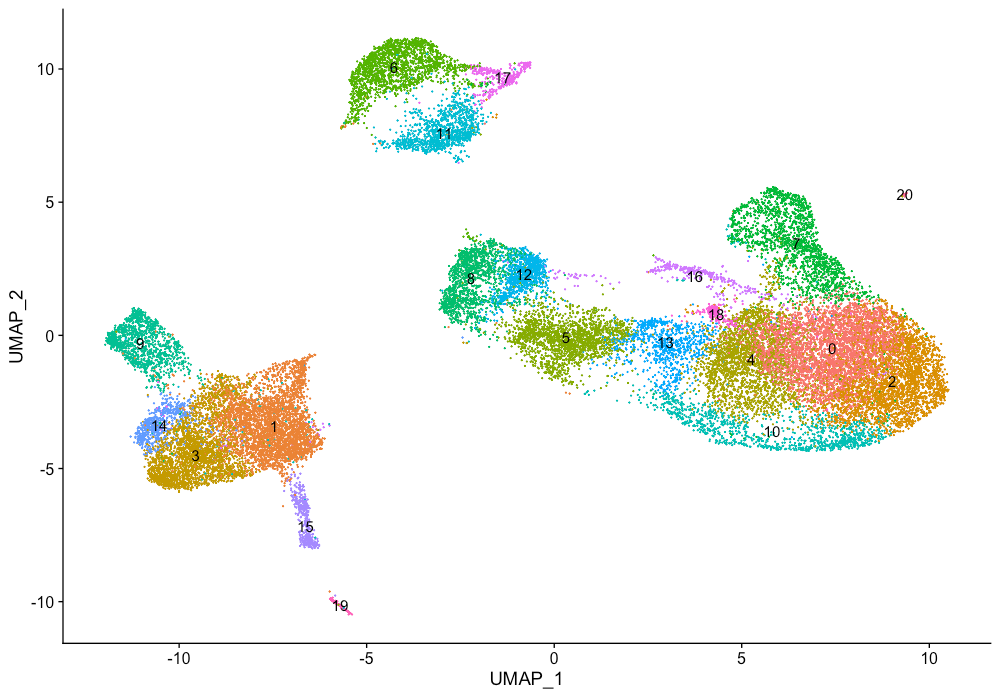
可以利用FeaturePlot()对细胞marker进行展示:
利用Seurat的FeaturePlot()函数可以轻松地在UMAP可视化上探索已知的标记,进而确定簇的身份。同时我们可以使用RNA assay slot中的标准化计数数据,来访问所有基因的表达水平(而不仅仅是3000个高度可变的基因)。
# Select the RNA counts slot to be the default assay
DefaultAssay(seurat_integrated) <- "RNA"
# Normalize RNA data for visualization purposes
seurat_integrated <- NormalizeData(seurat_integrated, verbose = FALSE)同时还需要考虑簇与簇之间marker表达的一致性。例如,如果一个细胞类型有两个标记基因,而簇中仅表达了其中一个,那么我们就不能绝对地将该簇分配给该细胞类型。
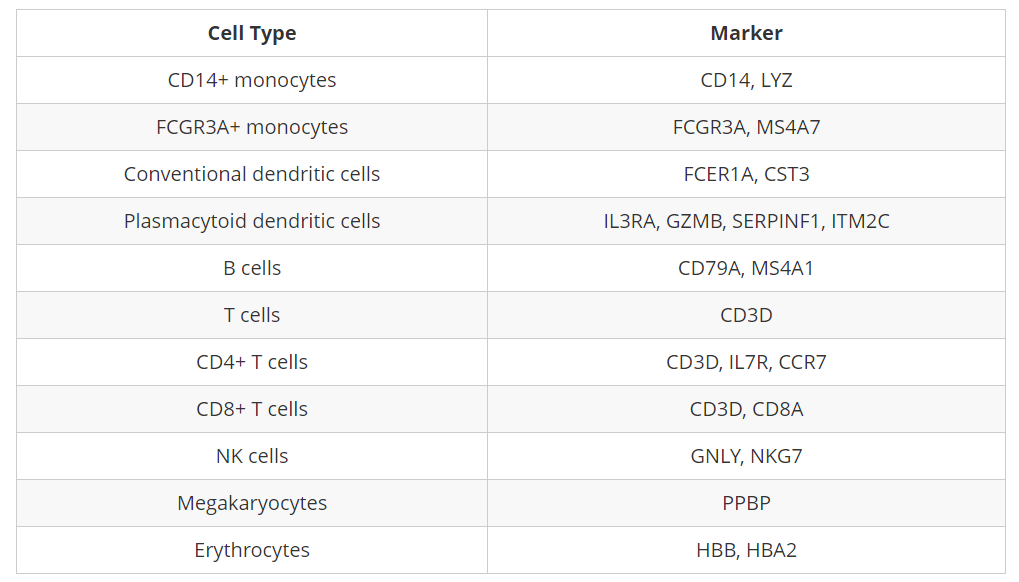
CD14+ monocyte markers
FeaturePlot(seurat_integrated,
reduction = "umap",
features = c("CD14", "LYZ"),
sort.cell = TRUE,
min.cutoff = 'q10',
label = TRUE)
从Marker表达来看,CD14+单核细胞似乎与cluster 1、3和14相对应。
FCGR3A+ monocyte markers
FeaturePlot(seurat_integrated,
reduction = "umap",
features = c("FCGR3A", "MS4A7"),
sort.cell = TRUE,
min.cutoff = 'q10',
label = TRUE)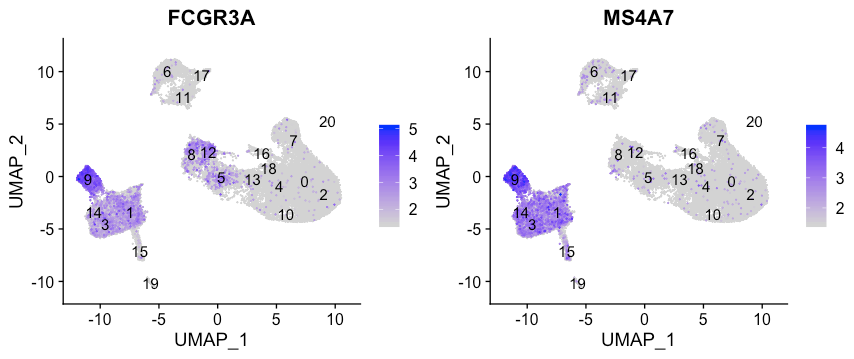
从Marker表达来看,FCGR3A+单核细胞与cluster 9对应。
Macrophages
FeaturePlot(seurat_integrated,
reduction = "umap",
features = c("MARCO", "ITGAM", "ADGRE1"),
sort.cell = TRUE,
min.cutoff = 'q10',
label = TRUE)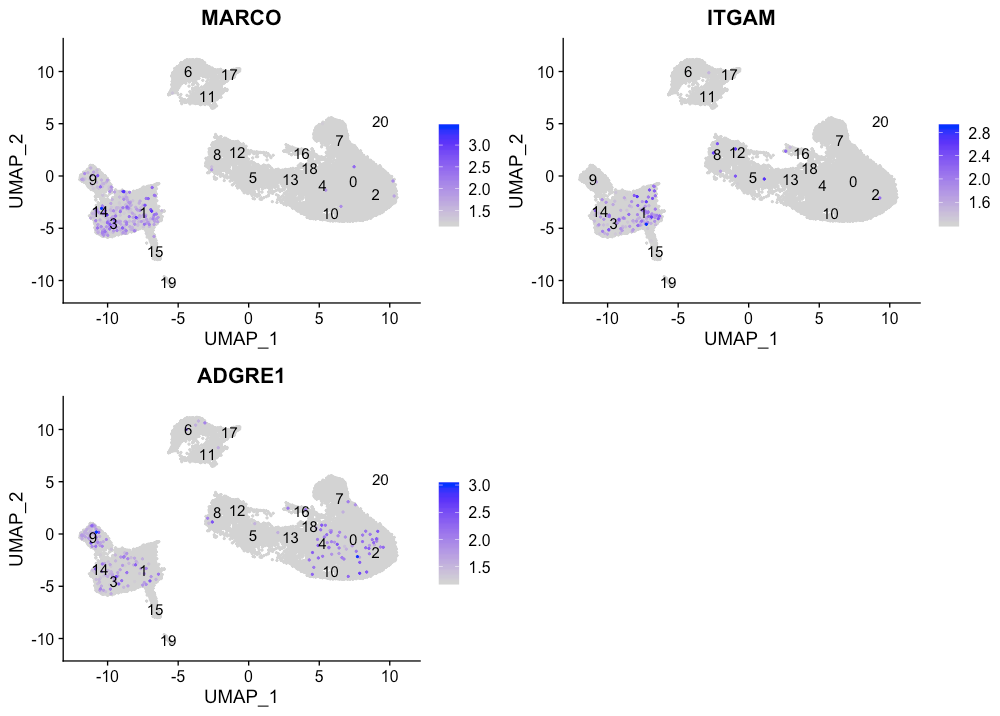
似乎没有cluster对应于巨噬细胞。
Conventional dendritic cell markers
FeaturePlot(seurat_integrated,
reduction = "umap",
features = c("FCER1A", "CST3"),
sort.cell = TRUE,
min.cutoff = 'q10',
label = TRUE)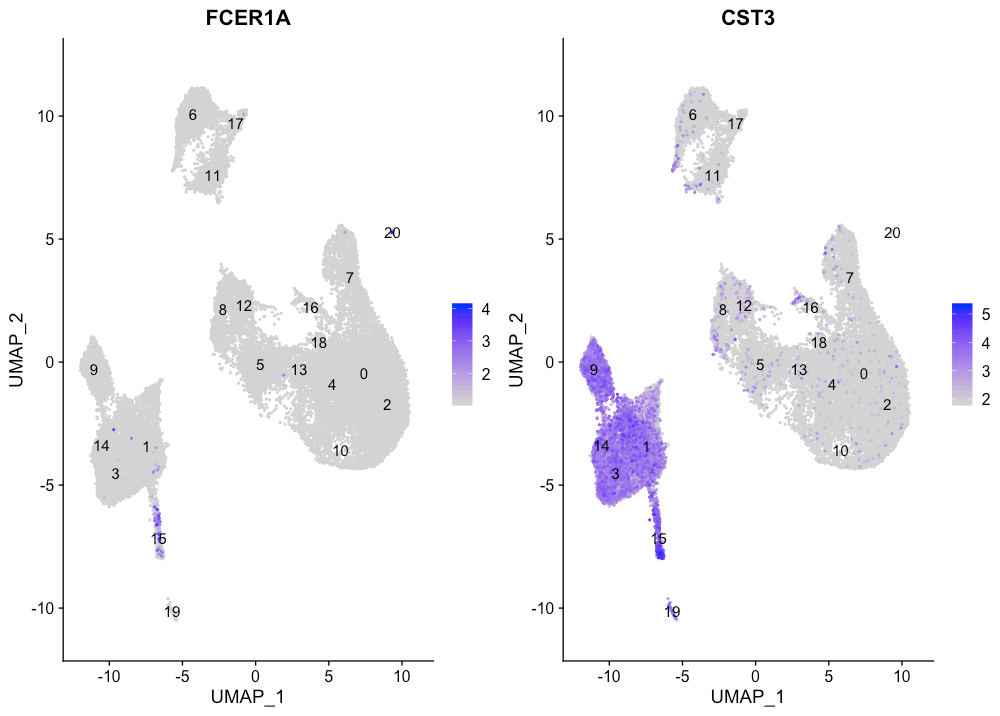
conventional dendritic cells(cDC)对应于cluster 15。
Plasmacytoid dendritic cell markers
FeaturePlot(seurat_integrated,
reduction = "umap",
features = c("IL3RA", "GZMB", "SERPINF1", "ITM2C"),
sort.cell = TRUE,
min.cutoff = 'q10',
label = TRUE)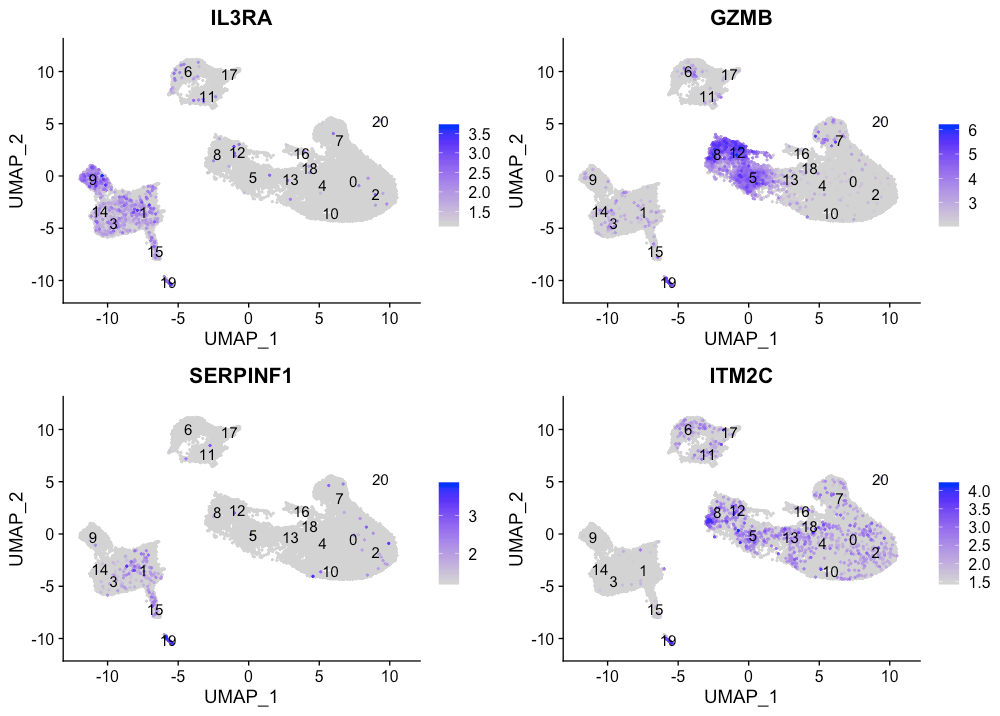
Plasmacytoid dendritic cells (pDC)对应于cluster 19。
NOTE: If any cluster appears to contain two separate cell types, it’s helpful to increase our clustering resolution to properly subset the clusters. Alternatively, if we still can’t separate out the clusters using increased resolution, then it’s possible that we had used too few principal components such that we are just not separating out these cell types of interest. To inform our choice of PCs, we could look at our PC gene expression overlapping the UMAP plots and determine whether our cell populations are separating by the PCs included.
上面的分析中还存着待解决的问题:
cluster 7和20的细胞类型标识是什么?
对应于相同细胞类型的簇是否具有生物学上有意义的差异?
这些细胞类型有亚群吗?
通过为这些簇鉴定其他标记基因,我们能否更相信这些细胞类型身份?
下一步将执行marker识别分析,该分析将输出簇之间表达差异显著的基因,能对上面的问题进行解惑。使用这些基因,我们可以确定或提高对簇/亚群身份的信心。