Nat. Commun. | 针对急性骨髓性白血病联合疗法的二分图网络模型
编译 | 任宣百
审稿 | 姜晶
本次报道的论文是来自赫尔辛基大学医学院的Jing Tang老师团队发表在nature communications上Bipartite network models to design combination therapies in acute myeloid leukaemia。文章提出了一个通过对患者相关药物反应数据的二分图网络建模来识别潜在的药物组合。在此项研究中选择性药物组合的药效和协同作用水平在体外治疗急性骨髓性白血病的三个细胞系中证实了与单药治疗的区别。本文中介绍了一种名义上的数据挖掘方法,通过组合疗法改善急性骨髓性白血病的治疗方案。

研究背景
联合治疗或使用协同药物的综合治疗可以通过针对同一或多个靶点,实现更有效、更安全的治疗效果。传统的方法没有考虑到体外药物筛选,解读癌症基因组学在表型上的功能影响,和理解它们在生物网络背景下的相互作用。即使有的网络重建来模拟疾病的生物机制,并根据分子数据预测药物组合的协同作用,但网络模型还没有系统地应用于患者数据,如患者来源样本的药物反应数据,以预测患者定制药物组合。本文中提出了一种网络药理学方法来预测基于BeatAML数据集的急性髓系白血病(AML)的潜在药物组合。通过利用体外药物筛选数据的二分图网络模型的药物联合策略。结合体外药物反应数据,直接获取患者癌细胞的个体表型,并通过网络建模,证明了药物和AML患者的相似性(图1)。
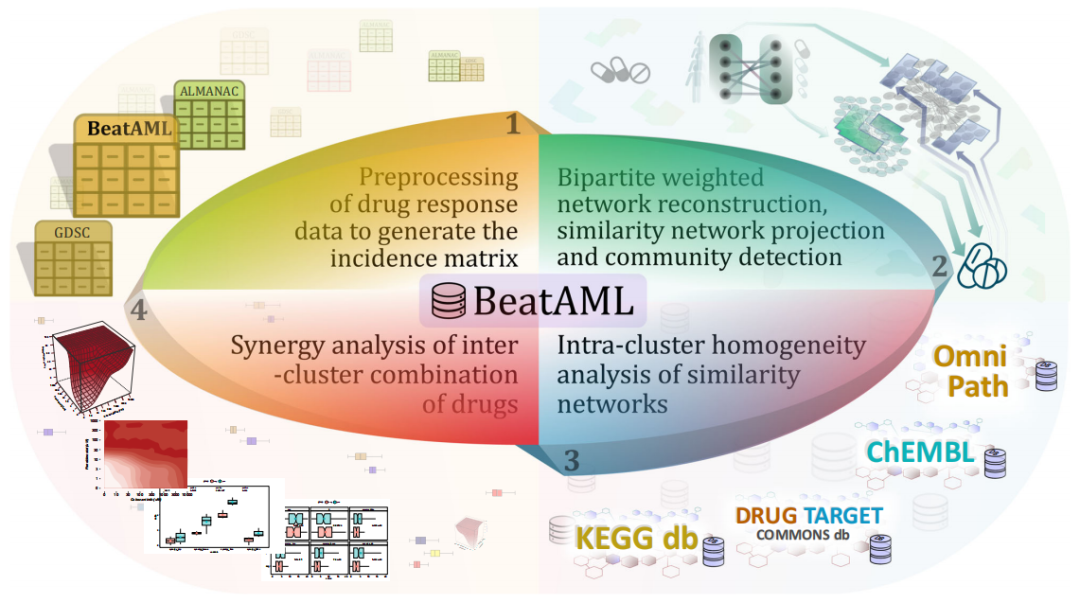
图1. 研究流程图。
实验步骤及结果
定义二分图网络的边权值
确定了用于二分图网络重建的药物-样本相互作用的权值。在药物敏感性分析中,这个值应该描述抑制肿瘤细胞最有效的化合物。再加上相对和绝对的半抑制浓度、RI值和AUC,作者计算了药物反应实验中的中位细胞活力。这些度量在正态性分布方面被评估为偏态和模态(图2),以在二分图网络中选择最佳的度量作为权重。中位数与AUC的关系有较高的正值(r-Pearson correlation coefficient~0.94)。中位数分布与半抑制浓度分布相比为单峰分布,与RI分布相反,与AUC分布相比更对称(非偏态)。
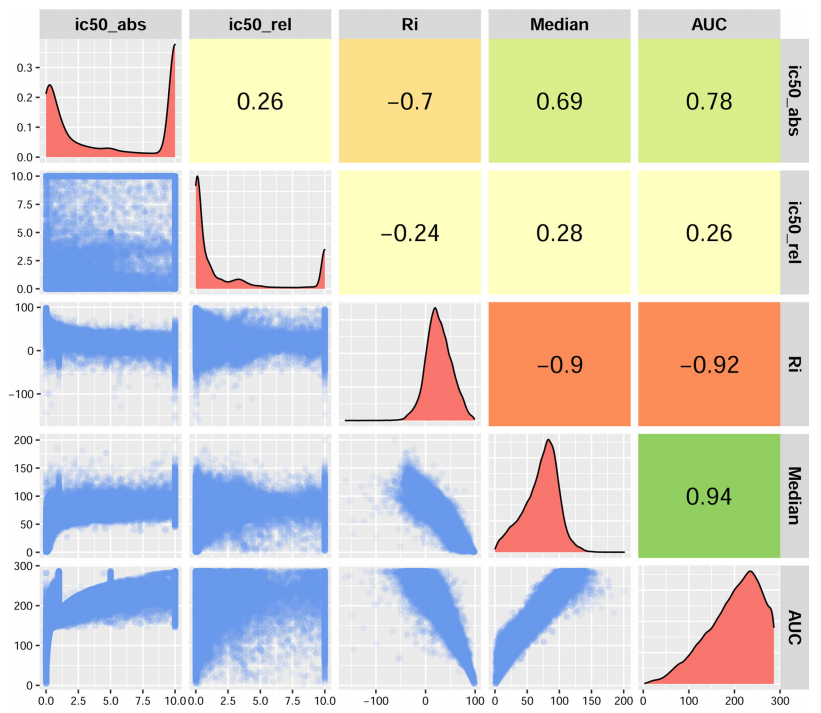
图2. Beat AML研究中药物反应实验的不同措施的比较
二分图网络分析
作者选择了列表删除策略来删除缺失值,并使用了两个变量的完整情况。构建患者样本和小分子的全平方子矩阵(没有缺失的条目)作为二分图网络的关联矩阵。下游分析是在一个由176个(88+88)节点和7744条边组成的无向加权大幅图上进行的(图3a)。最小-最大归一化边缘权值的分布显示为正偏态,表明细胞对大多数药物不高度敏感。GDSC数据集的无向加权重图由532个(266+266个)节点和70,756条边(图3)组成。最小值-最大值的分布已归一化在该数据集中,边缘权重也显示出正偏态(图3),再次表明大多数药物的药效较低。因此,探索最佳组合需要对药物-样本相互作用进行分类。根据图3所示的这些大图的投影,重建两个投影图,即患者相似网络(PSN)和药物相似网络(DSN),通过将加权关联矩阵相乘得到每条边。
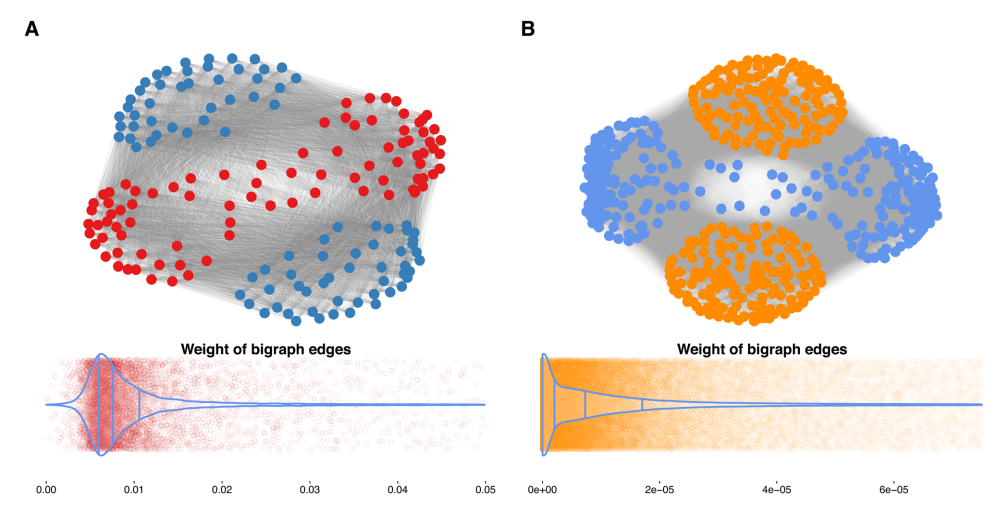
图3. 癌症数据集的大图
Beat AML数据集的PSN和DSN包含88个节点(图4左)和3828条边(图4右),而在GDSC投影相似度网络中,有266个节点和35,378条边。在图4中,节点大小越大,患者来源的样本越敏感,药物效力越强。
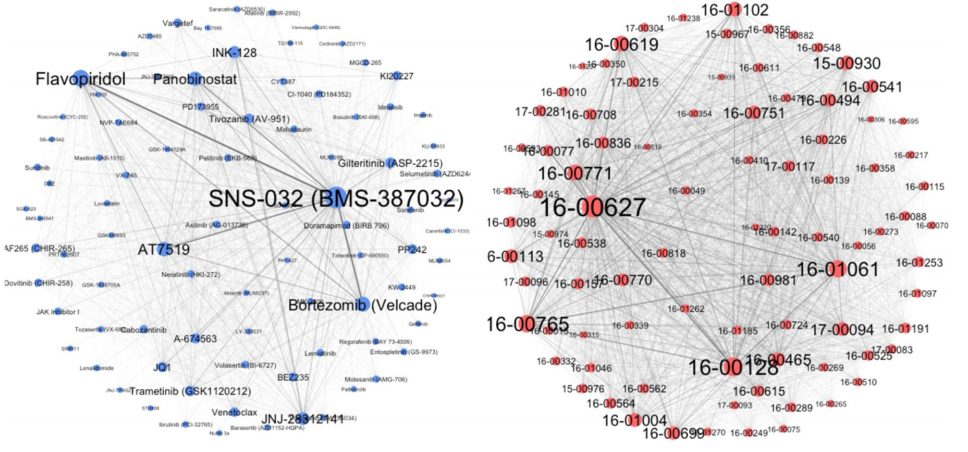
图4. BeatAML数据集的DSN和PSN
相似性网络的群内同质性分析
针对小分子,作者推测对细胞存活有相关影响的抑制分子往往具有相似的结构、目的和功能。因此,作者评估了SMILES结构的相似性,蛋白质靶标的类比,以及在DSN中检测到的集群与分子随机分组的生物途径。基于网络拓扑结构的随机分组和聚类之间的SMILES结构的Dice相似性分布存在显著差异(图5a)。网络集群内交叉点的中值相似度量明显超过了大量随机小分子对的相似值(图5b-d)(p-value<2.2e−16)。通过分析GDSC数据集(图5e-h)(p-value<2.2e−16)获得了可比性结果,表明方法也适用于基于细胞系的数据集的可重复分析。作者还比较RPKM值的调和平均相似性,聚类内患者的两两相似性显著超过了随机选择的患者(p -value<2.2e−16)(图5i)。对于CPM数据集,显示了Jaccard距离的分布,其中集群内的距离在统计学上低于随机组(p-value=4.655e−05)(图5j)。研究结果表明,聚类内的距离远低于随机分组(p-value=6.94e−08)(图5k)。
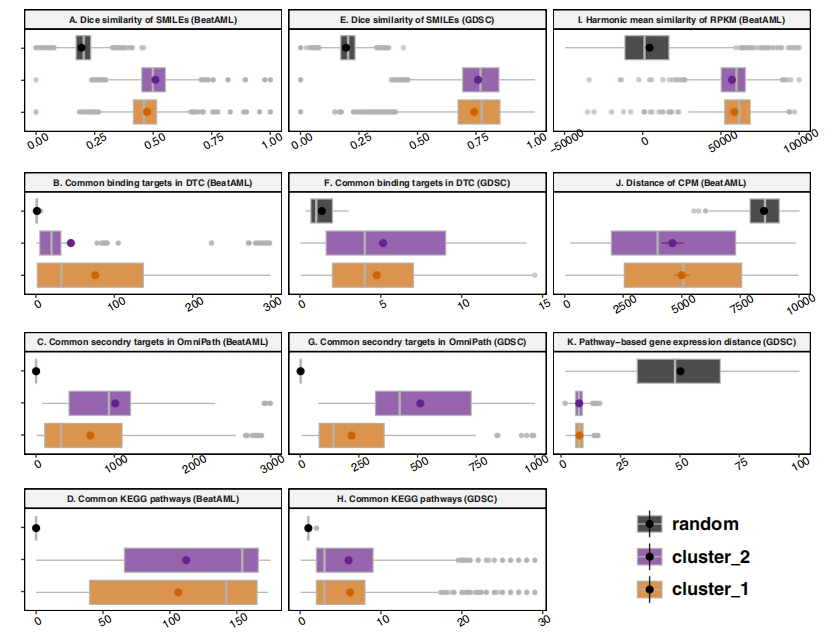
图5. Beat AML和GDSC的集群内同质性分析
BeatAML的研究也提供了AML的突变情况数据。在这里,作者使用了一个非良性基因突变的数据集来表征这两个患者样本的集群。如图6所示,这两组患者都表现出了不同的基因突变特征和基于频率的基因排名。
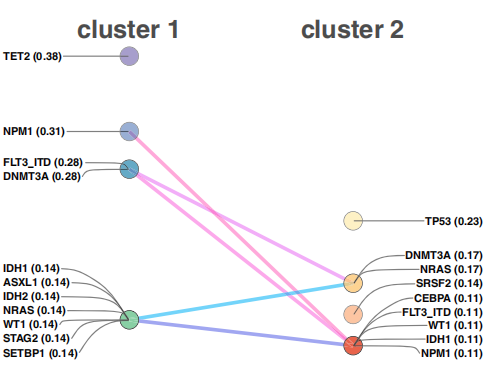
图6. Beat AML患者集群中频繁突变的基因
药物组合的群间设计策略
作者假设最佳的药物联合策略是从每个集群中选择一种药物,以阻断潜在的耐药机制和癌症复发。首先,作者检查了BeatAML和GDSC数据集中每个聚类的前5种药物的组合(基于细胞活力的中位数)是否能在DrugComb数据库中找到。图7显示了DrugComb中协同作用值的分布,突出显示了每个网络集群中底部和前五种药物协同作用的平均值。根据BeatAML和GDSC数据集的平均中位数(p-value=2.96e−02和p-value=3.56e−02),该分析揭示了前5种药物的组合具有相当高的潜力。
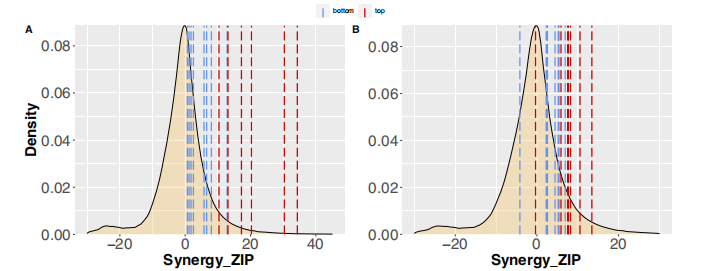
图7. 在DrugComb数据库中药物组合协同作用得分的分布
药物群间组合的协同作用分析
为了进一步验证利用网络建模预测协同药物组合的策略,作者重点研究了 ALMANAC数据集,其中有103种抑制剂的1,892,650个组合在60个细胞系上进行测试。图8a显示,阳性联合用药组的药物协同作用值明显高于阴性联合用药组。这一结果在所有类型的协同作用措施中都很明显,表明了使用集群间药物组合的策略的优越性。这些数据也表明作者提出的基于网络的模型识别具有生物样本相似效应的药物的效率。为了证明模型在预测特异性和稳健的药物组合方面的能力,作者对3种AML细胞系MOLM-16、OCI-AML3和NOMO-1的45种药物组合进行了实验验证。使用ZIP、Bliss、HSA和Loewe模型来评估协同作用的程度,对135个药物-药物-细胞系三联体的实验验证结果(图8b)。当将阳性评分作为协同作用程度的证据时,模型在阳性组中预测的药物组合被证实为更强的协同作用(图9)。总的来说,这些结果证明了基于网络的预测在各种实验设置和协同评分模型中的稳健性,以及基于网络的模型检测新的治疗组合的能力。
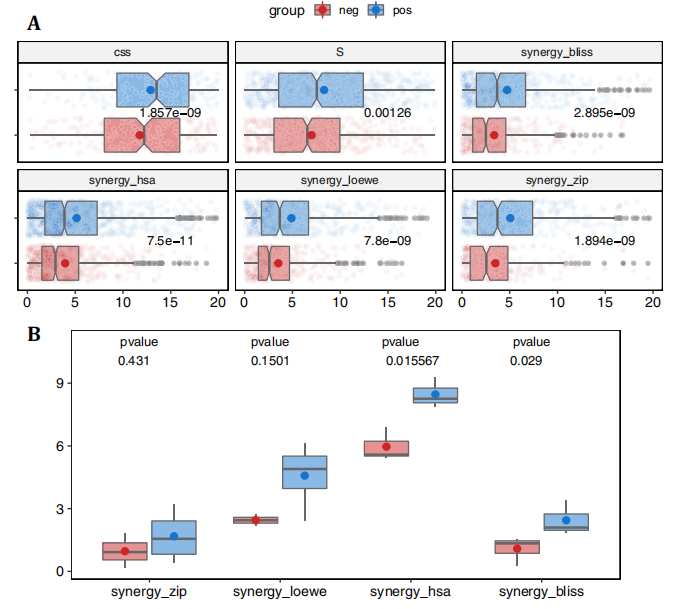
图8. 药物组合的协同作用
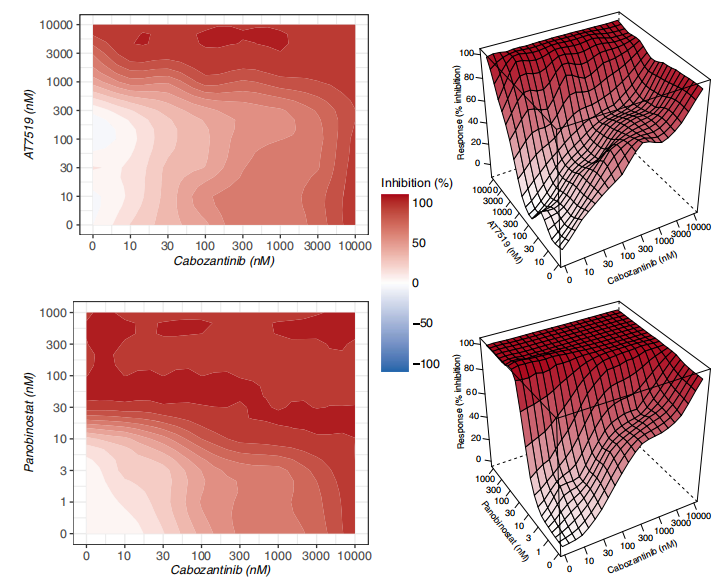
图9. 在阳性组中发现的最主要的协同作用药物组合
总结
在本研究中,作者重新考察了使用图论进行数据挖掘的药物筛选结果中的名义变量的分析,即药物名称和样本身份,称之为名义数据挖掘方法。首先考虑数据质量控制,如离群值检测、异常值处理、生物和技术复制。同时,证明了中位数可以代表比较网络重建药物功能的一个适当的权重评分。通过加权网络投影提供两个相似度网络来检测网络群落的拓扑结构。该方法不需要训练数据集来预测药物组合,仅使用药物反应数据就足以进行预测,而不需要整合生化谱的先验知识。
参考资料
Mohieddin Jafari, Mehdi Mirzaie, Jing Tang, et al. Bipartite network models to design combination therapies in acute myeloid leukaemia. Nature Communications, 2022, 13: 2128.
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集