
基因与疾病百年研究进展
来源:生信人 本文约3900字,建议阅读7分钟
本文为你介绍一篇生信分析者充电的杂志文章。

今天我们给大家介绍一篇题为“100 Years of evolving gene–disease complexities and scientific debutants”的Briefings in Bioinformatics(IF:8.9)杂志文章。
摘要
“基因”这个词已经有100多年的历史了,并在几个科学方向上不断发展。随着技术的不断进步,基因组学领域发生了重大变革,尤其是当涉及到诸如三重编码开发、基因数命题、基因图谱、数据库、基因-疾病图谱、人类基因和遗传疾病目录、CRISPR/Cas9、大数据和下一代测序等领域时,在这篇文章中,该研究团队介绍了基因组学从豌豆植物遗传学到人类基因组计划的进展,并重点介绍了分子、技术和计算的发展。
基因组和表观基因组的研究为人类疾病的发生和发展提供了基础,包括染色体疾病、单基因疾病、多因素疾病和线粒体疾病。世界卫生组织对所有人类疾病进行了分类、标准化和维护,当时许多学术和商业在线系统正在共享有关基因的信息,并与相关疾病相关联。为了有效地了解这些生物数据的丰富性,迫切需要生成适当的基因注释库和资源。
该研究团队的重点是全世界有多少基因疾病数据库,哪些来源是真实的、及时更新的,并推荐用于研究和临床目的。在本研究中,该研究团队讨论并比较了43个这样的数据库和生物信息学应用程序,这些应用程序使用户能够连接、探索并在可能的情况下下载基因疾病数据。
背景介绍
基因研究:帮助为每个人提供量身定制的解决方案。人类基因组序列的变异性是负责人类发育和功能的生物代码的结果。人类基因组的大部分(约62%)由基因间区域组成,这些区域是基因组中位于基因之间的非蛋白质编码部分,过去被称为“垃圾DNA”,但过去几年的基因组研究揭示了与这些区域相关的功能,这表明基因组的每一部分都有一定的重要性。
基因间DNA还可能包括基因调控序列,如启动子、增强子和沉默子,这些序列尚未被鉴定。核糖核酸(RNA)是DNA的转录形式,信使RNA(mRNA)是RNA的蛋白质编码形式。非编码RNA,如转移RNA(tRNA)、微RNA(miRNA)、核糖体RNA(rRNA)和长非编码RNA(lncRNA),在细胞中扮演着从蛋白质翻译到基因调控的各种角色。
本文要点
1)真实和分类的基因疾病数据在生物研究和医疗保健的各个层面都具有非常重要的意义。
2)为了有效地挖掘基因组学和临床数据的财富,迫切需要开发合适的基因疾病注释库和资源。
3)需要很好地解决不断演变的基因疾病复杂性,以便更好地在全球范围内实施精确医学。
4)创新和强大的大数据平台对于通过智能分析和共享临床和基因组数据来发现新的治疗靶点和设计诊断工具来提高医疗质量是必要的。
基因学史:分子、技术和计算
1869年,在德国图宾根工作的瑞士生物化学家约翰·弗里德里希·米歇尔(Johann Friedrich Miescher)发现了DNA。在Miescher发现之前三年,格雷戈尔·孟德尔(Gregor Mendel)已经公布了他使用豌豆植物进行育种实验的结果,这些实验是在布尔诺(Brno)的修道院花园进行的,布尔诺是现在的捷克共和国的一个中欧城市。根据他在1865年进行的豌豆杂交实验,遗传性状被定义为某些可能导致性状差异的细胞内物质。颗粒遗传概念的建立激发了人们对这种材料的本质的思考。孟德尔在布尔诺的《自然科学学会学报》上发表的论文描述了他的假设,即遗传是由单位因素控制的,遗传学家今天称之为基因的实体。1909年晚些时候,孟德尔的一个学生威廉·约翰森(Wilhelm Johannsen)终于用“基因”这个词来描述遗传单位。从那时起,一百多年来,人们一直在探索基因的结构、定位、数量和功能。

从测序到精确的基因编辑
由于多核苷酸链中核酸的顺序决定了遗传信息,所以对DNA进行排序的能力对破译密码至关重要。随着电泳技术的发展,Southern blotting被发明来检测特定的DNA序列。然而,电泳只能分离不同大小的DNA片段,不能进行测序过程。在Sanger测序法的基础上,在DNA合成过程中加入末端链的双脱氧核苷酸,通过电泳进行测序。桑格测序虽然费时费力,但已经被广泛使用了40多年,并帮助科学家完成了人类基因组的第一个序列。双酶系统(硫酰化酶/荧光素酶)的链终止反应避免了繁重的电泳,并实现了实时测序。1955年,Arthur Kornberg分离出一种DNA复制酶--DNA聚合酶,这是迈向下一代测序(NGS)的一个重要里程碑。基于焦磷酸测序(一种光检测方法)的NGS以更简单的测序步骤和更少的时间消耗开启了高通量测序的新时代。最新一代的测序技术解放了对DNA扩增的要求,并实现了单分子测序(SMS),从而避免了与扩增相关的偏差和错误。在DNA模板通过一个核苷酸碱基连接到底物上之后,适当的荧光可逆终止子dNTP(所谓的“虚拟终止子”)被洗涤、成像一次、切割并循环到下一个。多年来,测序方案、分子生物学和自动化方面的创新提高了测序的技术能力,同时降低了成本。
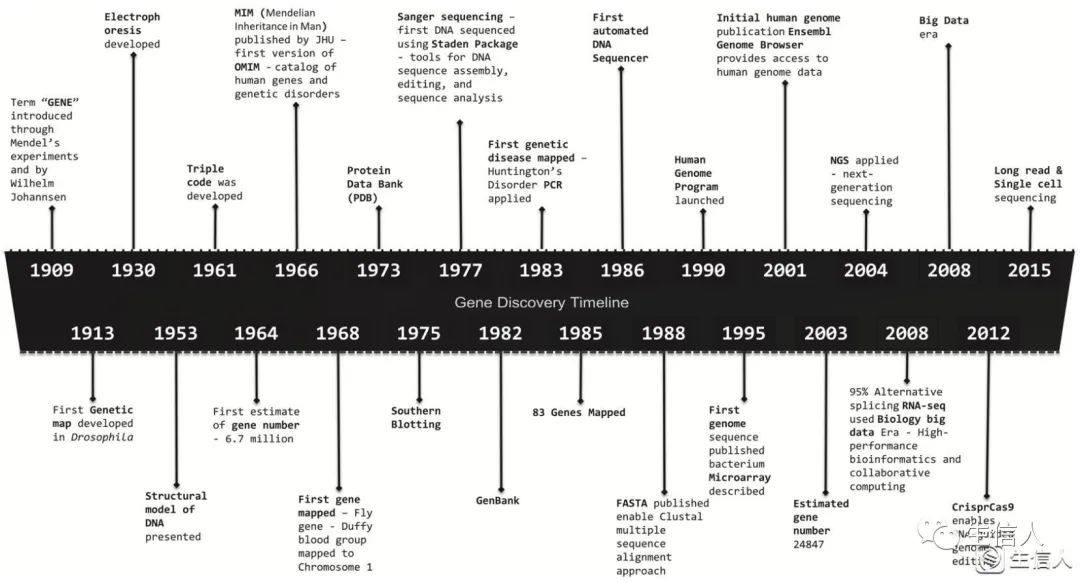
图1.基因发现时间表-介绍了从1909年到2015年基因组学领域的主要发现和发明
从基因组学到蛋白质组学
生物信息学始于蛋白质研究,因为人们普遍认为蛋白质携带遗传信息;1952年的噬菌体感染实验证明DNA是真正的遗传物质后,情况发生了变化。第一个生物信息学软件COMPROTEIN是由margaretdayhoff开发的,用Edman测序数据来确定蛋白质的一级结构。建立了第一个蛋白质数据库PDB。当研究人员确认了DNA的重要性后,研究的重点很快转向了基因测序。第一个专门用于分析Sanger测序读数的软件由Roger Staden于1979年发布,用于搜索Sanger凝胶读数之间的重叠;验证、编辑序列读数并将其链接到contigs;以及注释和操作序列文件。1985年,richardstallman发表了GNU的notunix(GNU)宣言,后来发展成为一种自由软件哲学,倡导用户可以自由运行、复制、分发、研究、更改和改进软件。在这一概念的影响下,随着各种基因测序数据的积累,世界各地的实验室和数据库合作建立了大量的公共数据库,目前仍在使用。随着2003年人类基因组计划的完成,这些基因数据库的基础数据源终于建立起来。第二代测序技术和相应处理工具的出现,使得整个生物信息领域的功能日益强大,标志着生物大数据分析时代的到来。但随着大数据的出现,出现了新的问题,比如现在大量可用的工具的产生,使得很难确定具体任务所需的工具。此外,越来越多的实验室需要分析大数据,即使他们不专门从事生物信息学研究。在未来,生物信息学的发展将集中在统一性和用户友好性上。
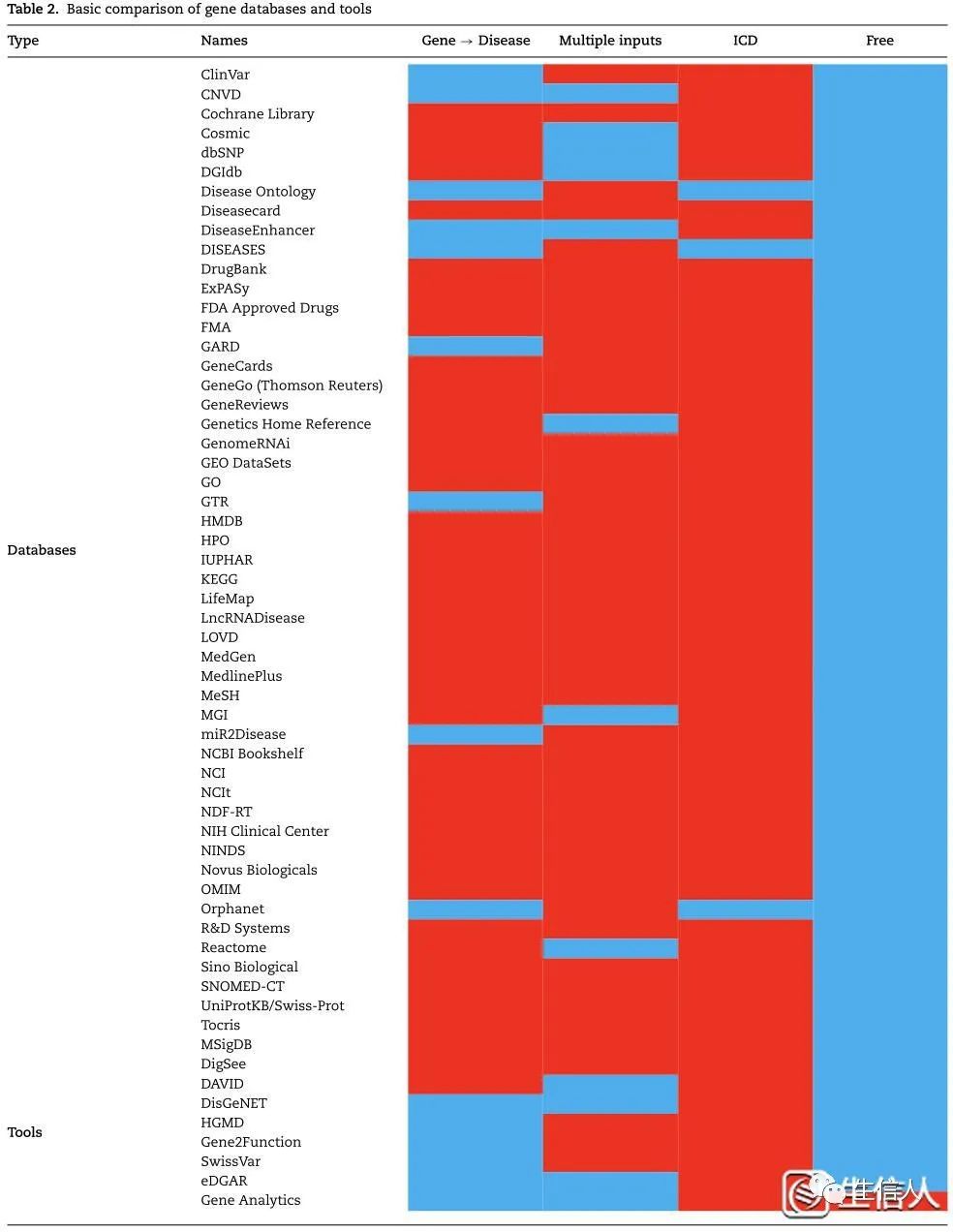
表2. 基因数据库和工具的比较(蓝色表示肯定,红色表示否定)
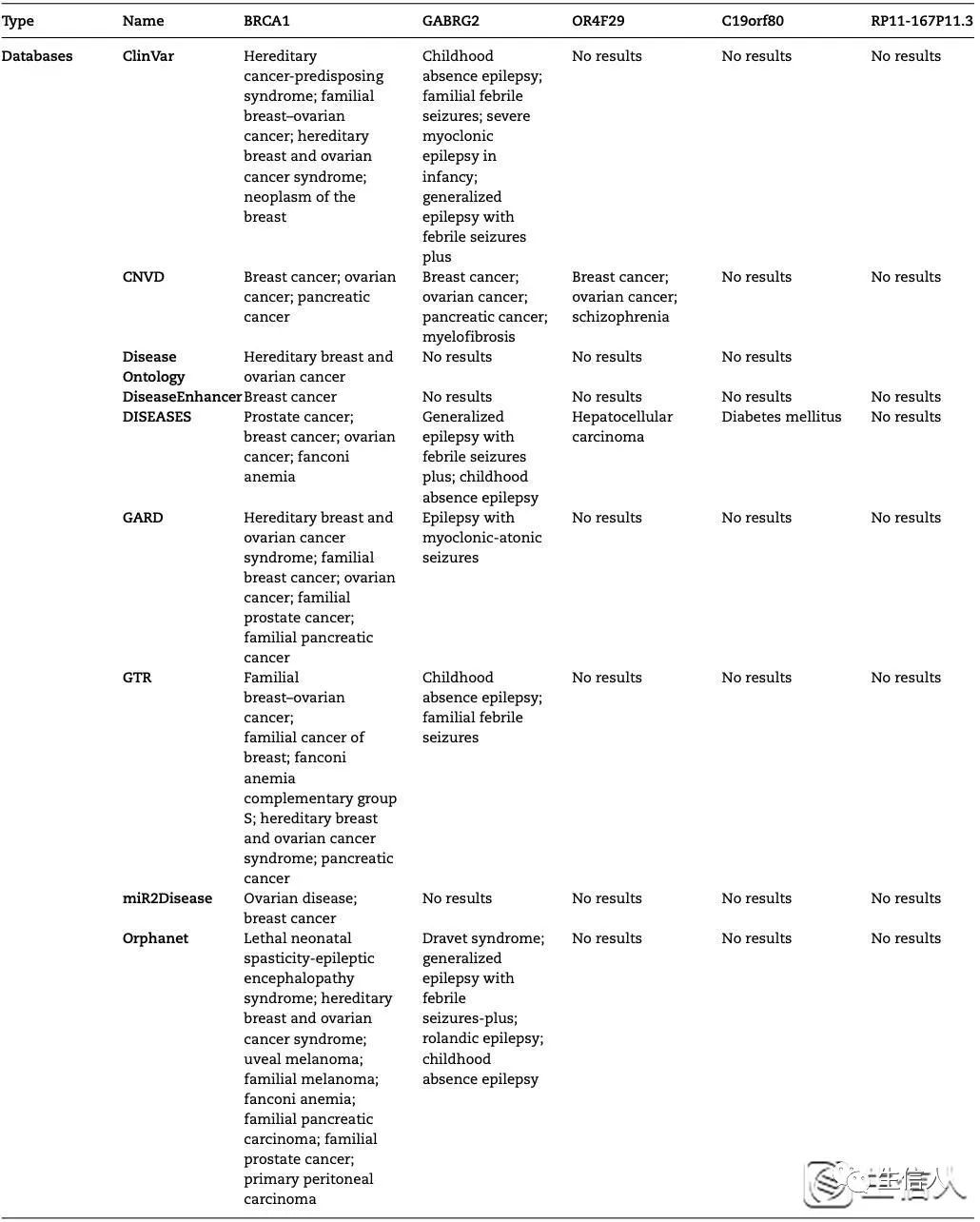
基因到疾病数据库和工具
随着高通量技术的发展,基因组规模科学正变得越来越普遍。这一领域的一个关键挑战是NGS数据的解释。科学家们面临着一项艰巨的挑战,那就是如何确定与其感兴趣的生物系统相关的候选基因。大多数情况下,研究人员只直接了解少数候选基因。许多复杂的疾病,如癌症,都是伴随着零星的基因组改变而发展起来的。这些改变可能导致功能性细胞问题或是惰性的。并不是所有的突变对发现它们的癌症类型都有同样的贡献。与癌症有因果关系的突变比例仍不清楚。尽管每个癌症基因组的独特变体数量可能非常高,但只有少数变体对肿瘤的发展至关重要。这就需要与综合知识库相联系的生物信息学工具。

图2. 基因到疾病数据库和工具
ClinVar
国家生物技术信息中心(NCBI)的ClinVar是一个免费提供的报告条件的变体档案。该数据库包括任何大小、类型或基因组位置的种系和体细胞变种。解释由临床测试实验室、研究实验室、位点特异性数据库、OMIM、GeneReviewsTM、UniProt、专家小组和实践指南提交。ClinVar目前持有超过158000份提交的解释,代表了超过125000个变量。数据库中的解释影响了26000多个基因,包括可能包含许多基因的结构变体;对于影响单个基因的变体,数据库中代表了近4800个基因。它允许用户输入一个基因名称,例如BRCA1,它将按名称检索所有相关的变异和相关的条件,以及他们的复查状态和上次复查的时间。在“帮助”页面中,他们声称自己的复习状态可以分为八个连续的等级,但他们不会根据这些等级显示成绩。尽管有一个选项可以根据评审状态进行筛选,但如果结果可以按照从高到低的等级等级来呈现,则会更有帮助。此外,他们的网站可以一次搜索一个基因,不能接受多个基因的输入,这就给研究人员留下了大量的工作来收集和整合信息。他们的结果主要来自三个不同的来源:临床试验、研究和文献。除了文献外,目前还没有足够的方法和系统来衡量临床试验结果的准确性,研究依赖于数据提供者。因此,有些信息可能模糊不清。
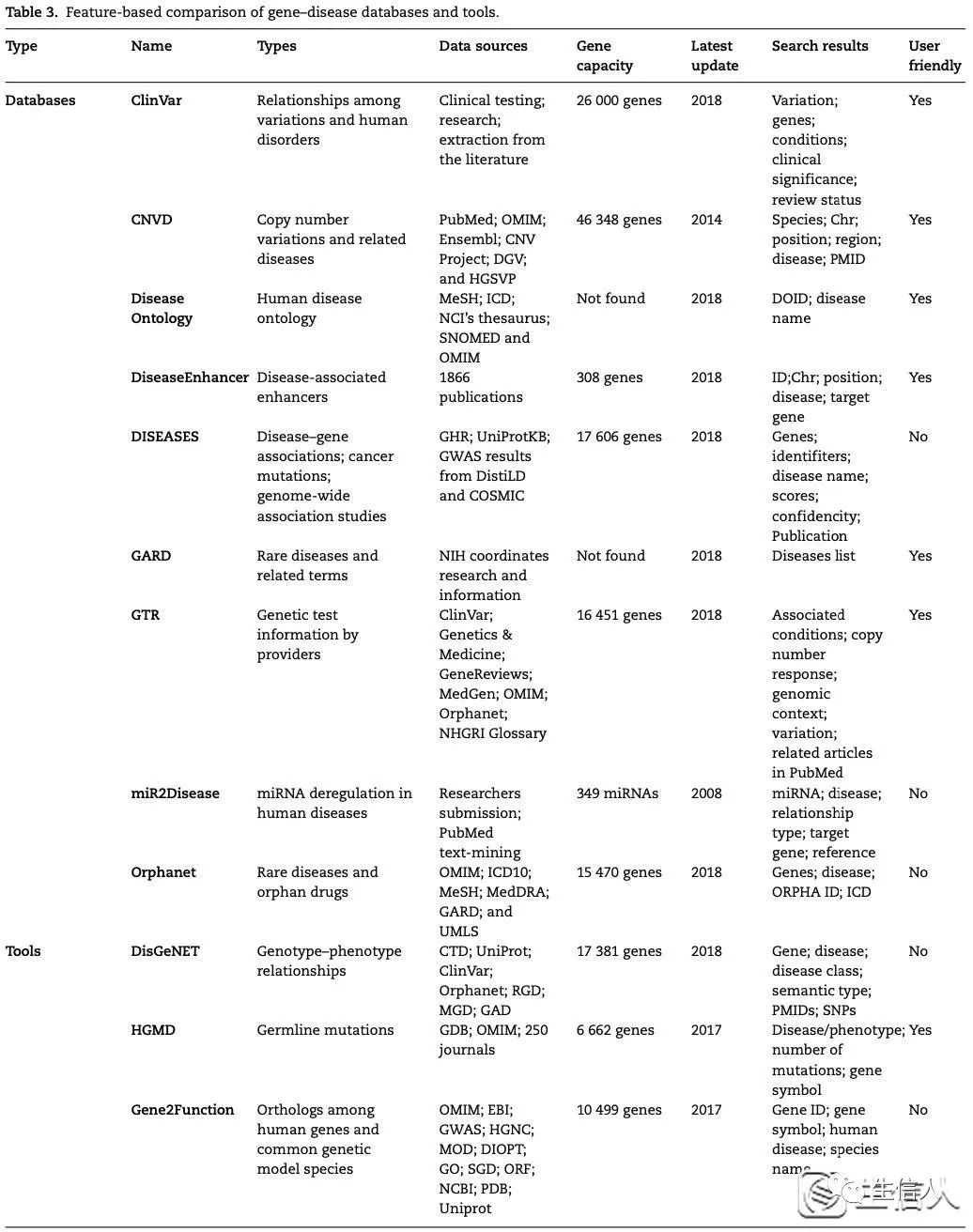
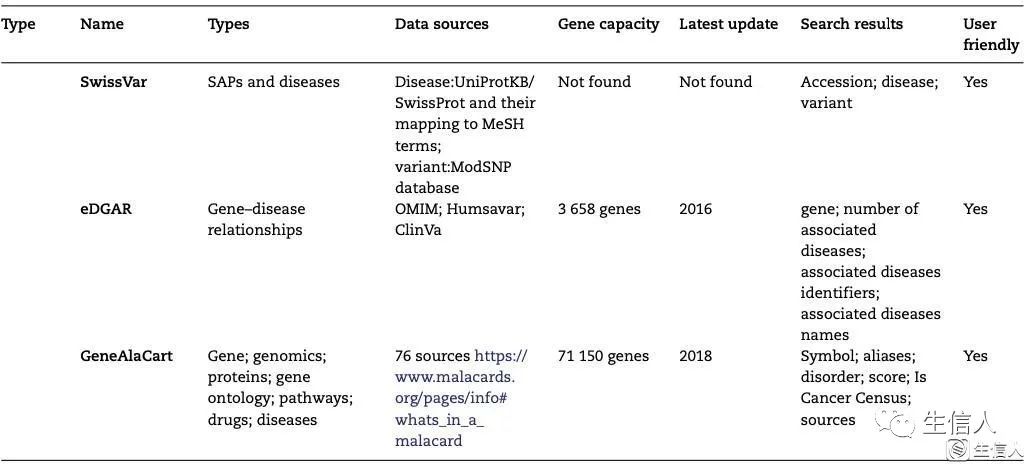
表3. 基于特征的基因疾病数据库和工具比较
疾病的拷贝数变异
疾病拷贝数变异(Copy Number Variations in Disease,CNVD)是一个系统而全面的拷贝数变异及相关疾病数据库,所有记录都是从CNV相关文献发表的实验数据中手工提取的。因此,CNVD是研究疾病相关拷贝数变异的可靠资源。它包含251697条记录,包含183219个CNV片段、844种相关疾病和46348个基因,这些基因是从2006年至2014年发表的文章中挖掘出来的。CNVD允许用户通过多种方式搜索数据库:通过基因名称、疾病名称、染色体位置或拷贝数变异区域。在查询结果中,每条记录都包含这样的信息:物种、染色体、CNV的起始和终止位置、相关疾病、CNV区域的基因以及源文章的PubMed ID。尽管CNVD是以拷贝数变异的基因为基础的,但结果仍然清楚地显示了基因名称和相关疾病。根据用户需要,可以批量下载,也可以单独下载。CNVD可以执行从基因到疾病以及从疾病到基因的双向搜索,促进基因注释。此外,它执行多基因搜索的能力给研究带来了极大的便利。但是,它不会对冗余的结果进行分组,这意味着如果相同的结果有不同的来源,它们将重复出现。此集成步骤需要用户自己完成,从而增加了意外的额外工作量。整个数据库完全基于CNV相关文章中发表的实验数据,因此数据量可能太小,无法考虑。
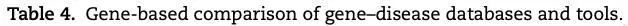
结论
尽管有许多重要的遗传学发现,但癌症等复杂疾病的遗传学仍很不清楚。随着对分子和基因组因素在疾病中发挥作用的逐渐了解,许多疾病(如癌症)有望得到更有效的医疗治疗。科学的方法将是对个体基因组进行个性化分析,从而在医疗保健领域产生一种新形式的预防性和个性化医学。基于基因的设计药物的可获得性,肿瘤分子指纹的精确定位,适当的药物治疗,预测个体对疾病的易感性,精神疾病的诊断和治疗,这些都是未来十年预计发生的许多变革中的几个。为了有效地理解丰富的生物数据,迫切需要生成合适的基因注释库和资源。