轻松在线差异基因/物种分析和可视化
7.1 limma用于转录组reads-count数据差异基因分析
做转录组差异分析,我们通常是基于reads-count数据,这个数据可以来源于Salmon的结果、STAR的结果或从网上下载的数据。
这个数据怎么获取,具体见下方的教程。
7.1.1 输入数据1:reads-count数据
现在假如我们已经拿到了8个样品的reads-count数据。点击下载ehbio_trans.Count_matrix.txt。
这是TAB键分割的文本文件:
第一列是基因名字,基因名可以是
ENSEMBL ID、Gene symbol或任意其它ID,但不允许有重复的名字。第一行除第一列
ENSG(第一列的名字可以是任意字符,不影响)外,其它列都是样本名字。样本名字建议只包括数字,字母和下划线。尤其是不能有-,不能以数字开头,最好下划线都不要有。数据矩阵中的其它内容都是整数。
从第二行起,每一行代表一个基因在每个样品里面的原始
reads-count值。
## untrt_N61311 untrt_N052611 untrt_N080611 untrt_N061011
## ENSG00000227232 13 25 23 24
## ENSG00000278267 0 5 3 4
## ENSG00000241860 3 11 1 5
## ENSG00000279457 46 90 73 49
## ENSG00000228463 5 4 13 6
## ENSG00000237094 0 16 7 2
## trt_N61311 trt_N052611 trt_N080611 trt_N061011
## ENSG00000227232 12 12 22 22
## ENSG00000278267 2 4 3 1
## ENSG00000241860 3 2 0 2
## ENSG00000279457 52 46 89 31
## ENSG00000228463 5 0 11 4
## ENSG00000237094 1 3 3 27.1.2 输入数据2:样本分组信息 (sampleFile)
既然做差异基因分析,通常是2组或多组之间的比较。如果是多组,也是两两组之间的组合比较。在上面的reads-count矩阵中只有样本名称,但不知道哪些样本是来源于一个生物处理组的,如对照组或处理组等。所以需要提供这样一个文件,指示样本的属性信息,如所属组信息。这里有一个示例数据,sampleFile2.txt:
## conditions individual sizeFactor SV1 SV2 SV3
## untrt_N61311 untrt N61311 1.0211325 -0.101 -0.494 -0.316
## untrt_N052611 untrt N052611 1.1803986 0.018 -0.170 0.588
## untrt_N080611 untrt N080611 1.1796083 -0.429 0.376 -0.089
## untrt_N061011 untrt N061011 0.9232642 0.535 0.241 -0.176
## trt_N61311 trt N61311 0.8939275 -0.125 -0.496 -0.366
## trt_N052611 trt N052611 0.6709229 0.036 -0.151 0.591需要注意的有一下几点:
样本分组信息
sampleFile文件至少需要有2列。第一列表示样本名字,需要与
reads-count文件中的第一行中的样本名字能对应上。顺序没有关系。大小写要一致。比如在sampleFile中样本名写做untrt_N61311,而在reas-count文件中写做UNtrt_N61311或untrt N61311,大小写不一致是不对的。一定注意是完全一致。第二列表示样本分组信息。示例中前4个文件属于
untrt组,后4个文件属于trt组。一个组的样本不要求必须挨着。样本的组名不要有除数字,字母和下划线之外的字符。其它列都是样本的属性信息,可有可无。有时也会用到,后面会有例子演示。
7.1.3 导入数据,设置参数
表达矩阵通常比较大,在1M左右或更大,直接粘贴进入浏览器的文本域会导致浏览器变卡。这不是网站速度慢的问题,是浏览器自身问题,不适合承载太大的数据。这里建议用上传的方式,免费注册一个账户,按下图所示上传数据。
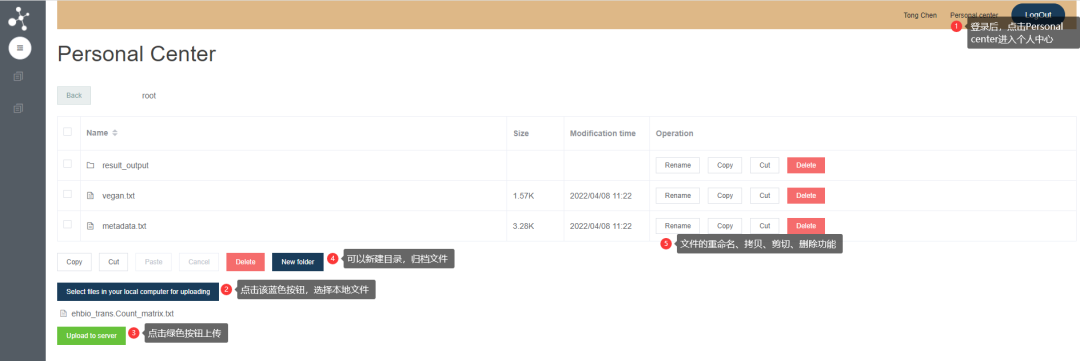
数据上传好之后,需要刷新工具页面,才可以看到新上传的数据。选择相应的数据,如下图
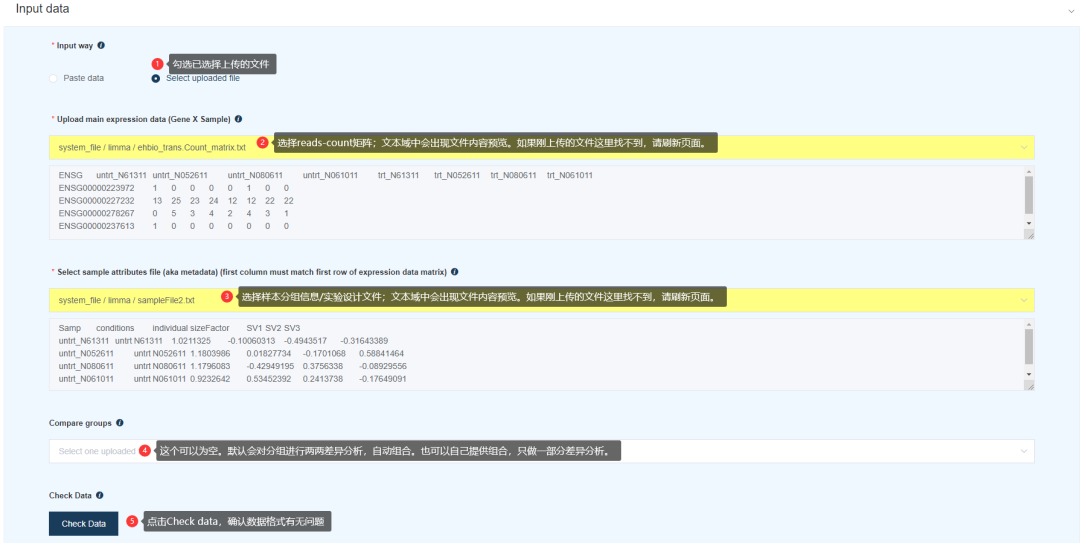
依次设置参数:
Input way设置为Select uploaded fileUpload main expression data (Gene X Sample)选择上传的reads-count文件Select sample attributes file (first column must match first row of expression data matrix)选择上传的sampleFile2.txt文件选择
Expression data type为Raw reads count选择
Group为conditions(sampleFile2.txt中表示分组的列)
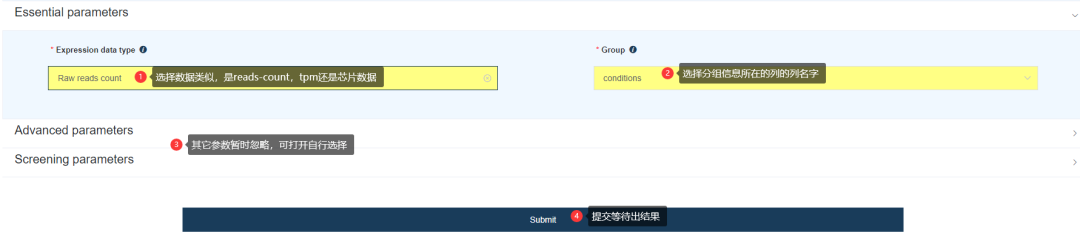
其它参数可以暂时先不管。
点击提交,获得结果。
7.1.4 差异分析结果报告
默认输出一个结果文档,1个大章节,2个小章节,分别是整体丰度图谱比较和差异分析结果。点击各个子章节可以展开查看具体内容。

7.1.4.1 1.1 所有样品丰度图谱展示 (聚类热图和PCA)
1.1.1
是样品表达图谱聚类热图,一幅下三角热图,一幅交互式热图。展示样本之间的相似性关系。标准化后的表达值也可从这下载。该工具主要定位是差异分析,可视化也有一些,不过都不能调整参数。如果需要进一步可视化,可以基于这里提供的表达数据,通过BIC的其它工具进行进一步可视化分析。后面我们会讲到。
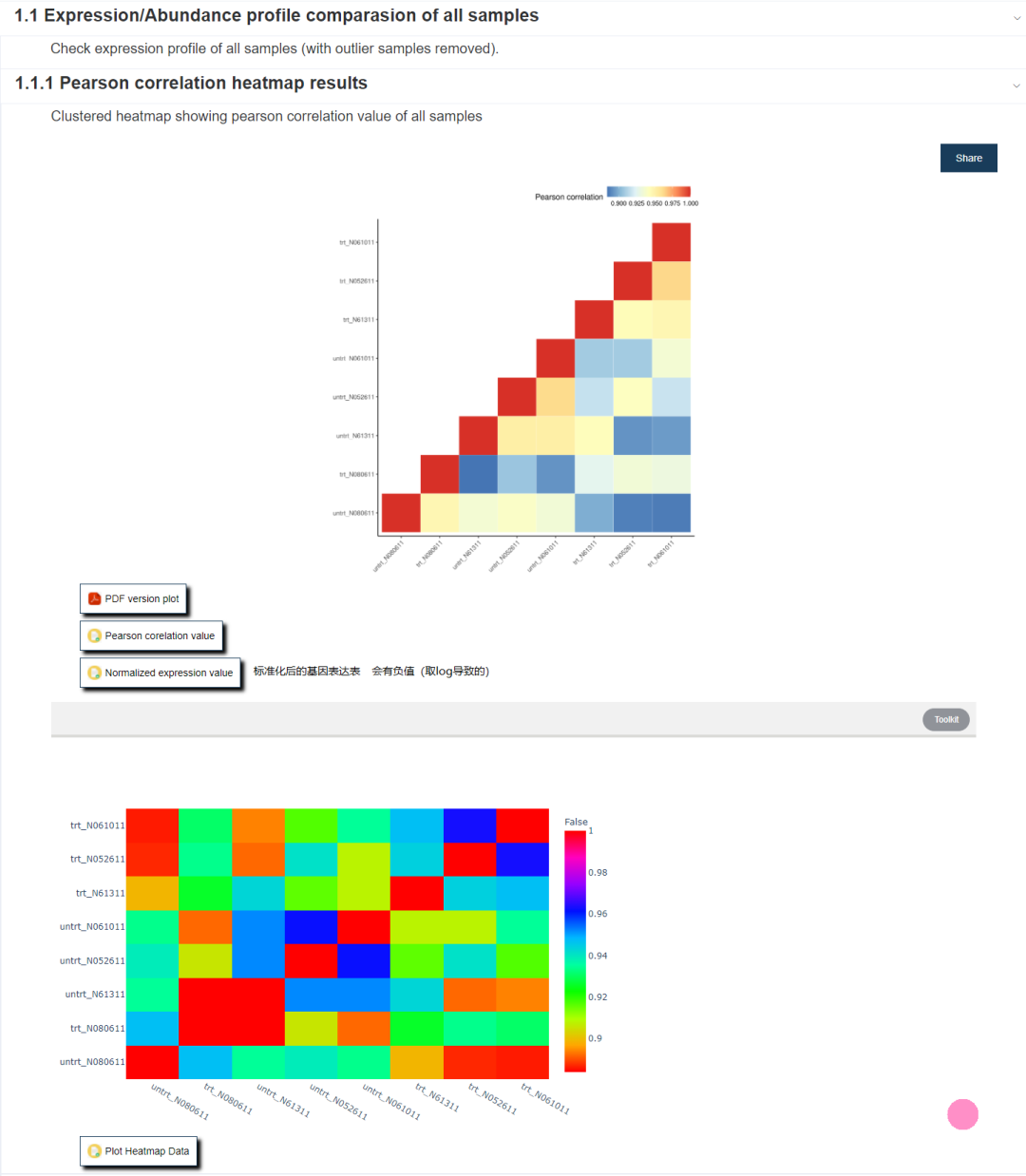
Figure 7.3: 点击理解图形解读 1. 这个热图上面的树是根据系统发育关系画的吗? 2. 图形解读系列 | 给你5个示例,你能看懂常用热图使用吗?
1.1.2 是样本表达图谱的PCA图谱绘制(静态图和交互图),在PC1轴上样品按处理和对照分成2簇,在PC2轴上,样品的分布与个体来源相关(这里是一个信号,我们后面会提到,感兴趣的可以先看下高通量数据中批次效应的鉴定和处理 - 系列总结和更新)。默认出图不太美观,数据可以下载下来,用我们的PCoA工具进行进一步绘制。
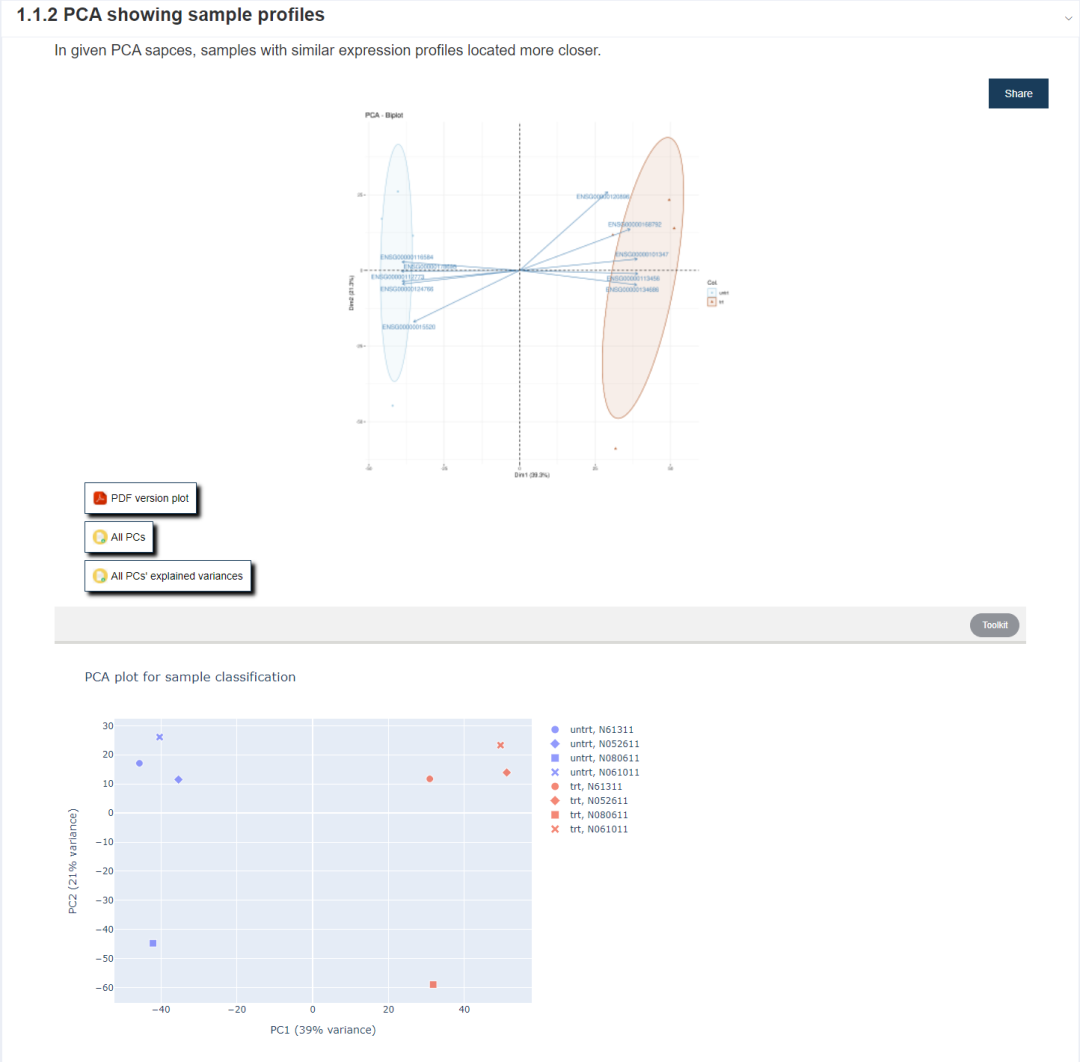
Figure 7.4: 点击理解图形解读 1. 一文读懂PCA分析 (原理、算法、解释和可视化) 2. 一文学会PCA/PCoA相关统计检验(PERMANOVA)和可视化
7.1.4.2 1.2 差异分析结果
因为只有一个比较组,所以只有1.2.1的结果;如果有多个比较组,还会有1.2.2 DE analysis results (A vs B), 1.2.3 DE analysis results (A vs C)等。
主要展现的是交互式差异火山图,鼠标悬浮可直接查看差异基因。火山图的数据也可以拿下来用BIC绘图平台定制绘制。
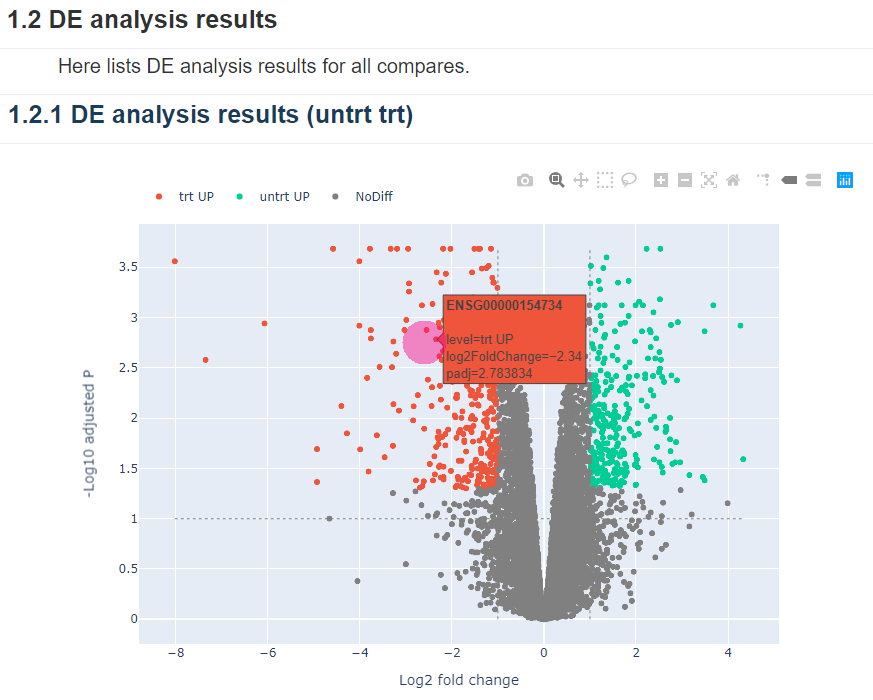
Figure 7.5: Volcano plot | 别再问我这为什么是火山图
差异分析的表格数据可下载用于进一步地分析和可视化。如下包括绘制火山图的数据、上调基因的表达矩阵、下调基因的表达矩阵、差异基因的列表等。

往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集