
ChatGPT启发,谷歌DeepMind预测7100万基因突变!AI破译人类基因遗传密码登Science

新智元报道
新智元报道
【新智元导读】AlphaFold之后,谷歌DeepMind再次震撼发布AI模型AlphaMissense,成功预测7100万「错义突变」,有望攻克人类遗传学难题。
蛋白质预测模型AlphaFold在AI界掀起海啸级巨浪后,Alpha家族又迎来新贵。
今天,Google DeepMind发布了全新AI模型——AlphaMissense,能够预测出7100万「错义突变」。
具体讲,AlphaMissense成功预测出的89%「错义突变」中,57%是致病性,32%是良性的。

论文地址:https://www.science.org/doi/10.1126/science.adg7492
而仅有0.1%的变异,能被人类专家确认。
为了研究人员更好了解其可能产生的影响,谷歌还将这份千万级「错义突变」所有目录公开。
一直以来,发现根本病因是人类遗传学面临的最大挑战之一。
而错义突变是可以影响「人类蛋白质」功能的基因突变,会导致囊性纤维化、镰状细胞贫血、癌症等疾病。
AlphaMissense的诞生展示了AI在医学领域,特别是在遗传学中的巨大潜力。
它对于理解遗传变异与疾病关系,开发针对性的药物治疗等都具有重要意义。
继AlphaFold之后,AlphaMissense或将成为足以改变世界的AI,有望攻克人类遗传学难题!
如果把DNA想象成一种语言,那么一个字母的替换就可以改变一个单词,并完全改变句子的意思。
在这种情况下,DNA的改变会导致氨基酸的变化,从而影响蛋白质的功能。
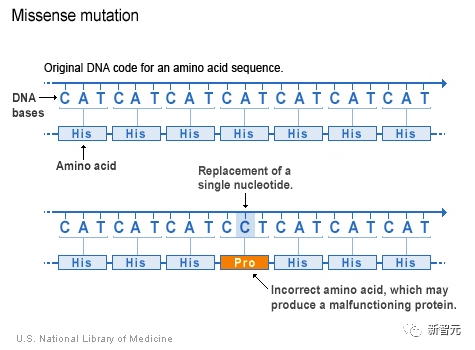
而普通人身上携带的错义突变超过9000多种。
一般而言,这些错义突变大多是良性的,对人体几乎没有影响。但其余少数则具有致病性,会严重破坏蛋白质的功能。
错义突变可用于罕见遗传病的诊断,因为少数甚至单个错义突变就可能直接致病。
此外,它们对于研究复杂疾病(比如ii型糖尿病)也很重要,这类疾病可能是由多种不同类型的基因变异共同引起的。

因此,对错义突变进行分类是了解哪些蛋白质变化可能导致疾病的重要一步。
在已出现的人类400多万个错义突变中,只有2%被专家标注为致病性或良性。
这仅占所有可能的7100万个错义突变的0.1%左右。
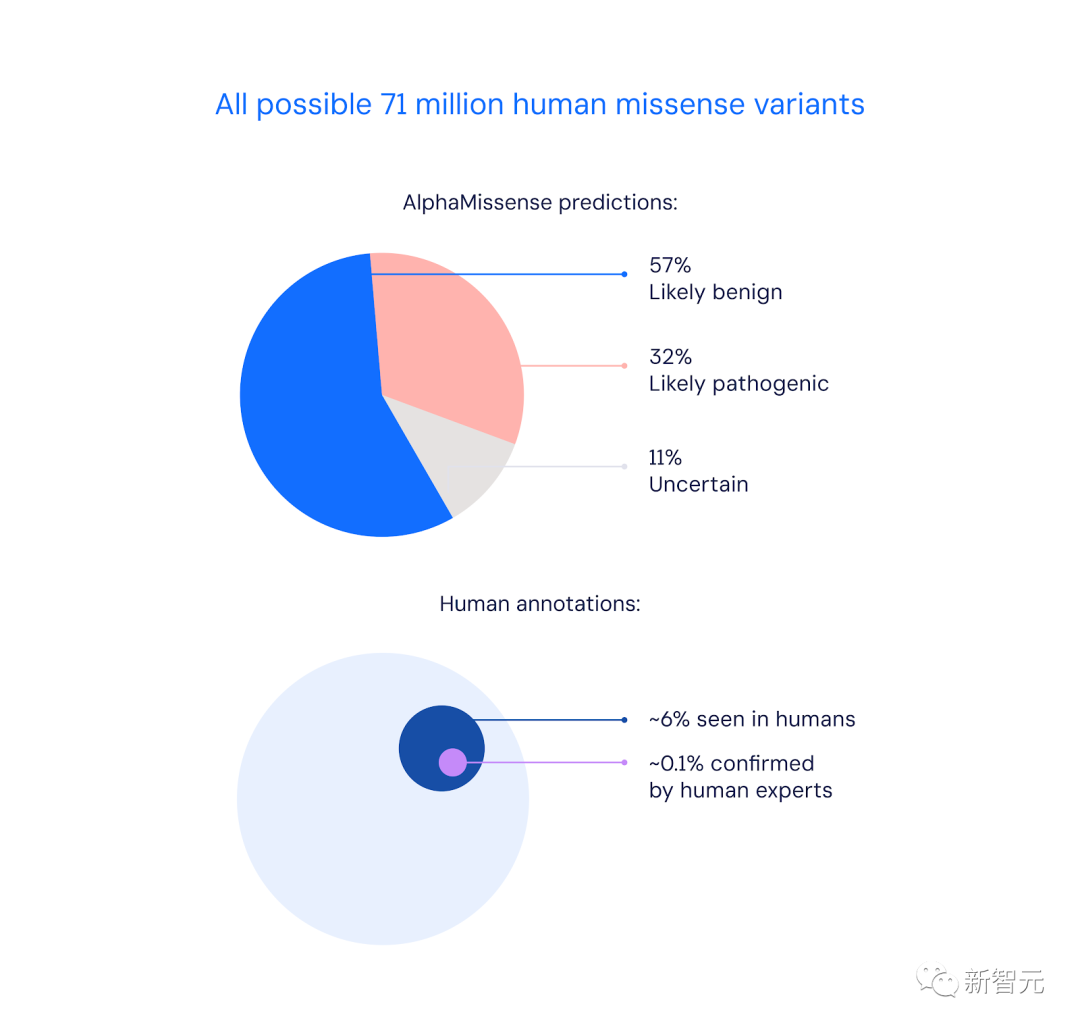
其余的突变因为缺乏相关影响的实验或临床数据,被归类为「意义不明的突变」。
但有了AlphaMissense,我们得到了迄今为止最清晰的突变影响图像:
AlphaMissense可以对89%的突变进行分类,其阈值在已知疾病突变数据库中的精确度为90%。

AlphaFold、AlphaFold 2自发布以来,已经从氨基酸序列预测了科学界已知几乎所有蛋白质的结构,超过2亿+蛋白质。
对此,谷歌研究人员基于AlphaFold(以下简称AF),对模型进行改编,由此可以预测改变蛋白质单个氨基酸的错义突变的致病性。
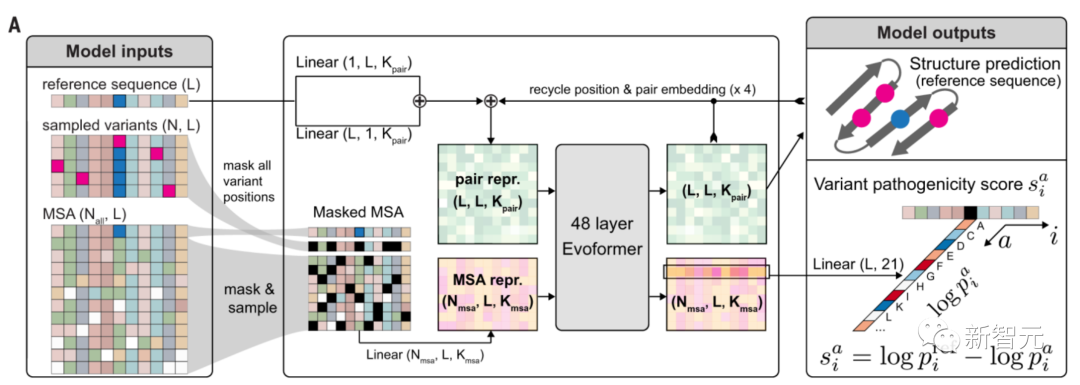
简单讲,AlphaMissense整个运作原理是:将一个氨基酸序列作为输入,并预测序列中给定位置所有可能的单一氨基酸变化的致病性。
为了训出AlphaMissense模型,需要分两阶段进行:
第一阶段
训练一个与AF一样的神经网络。这种神经网络的灵感来自像ChatGPT这样的大模型。
通过预测多重序列比对(MSA)中随机位置掩码的氨基酸身份,能够进行单链结构预测,以及蛋白质语言建模。
研究人员对AF进行了一些小的架构修改,并增加了蛋白质语言建模的损失权重,同时仍然实现了与AF相当的结构预测性能。
在预训练之后,掩码语言建模头已经可以通过计算参考氨基酸和替代氨基酸概率之间的对数似然比,来用于变异效应预测,正如MSA Transformer和进化比例建模(EMS)中所做的那样。
事实证明,这些神经网络擅长预测蛋白质结构和设计新蛋白质,尤其对变异预测很有用,因为它们已经知道哪些序列是可信的,哪些不是。
第二阶段
这个阶段,研究人员对模型在人类蛋白质上进行微调,并为MSA第二行中设置突变序列,增加变异致病性分类目标。
然后,按照按PrimateAI的方法,去标注人类和灵长类群体这种的突变。
常见的突变被视为良性,从未见过的突变被视为致病性突变。
一旦模型开始在验证集上过度拟合(2526个ClinVar变异,每个基因良性和致病性变异数相等),研究人员就停止训练。
不过,AlphaMissense不会预测突变后蛋白质结构的变化,或对蛋白质稳定性的其他影响。
而是,它利用AlphaFold对结构的「直觉」来识别蛋白质中可能发生的致病突变。
具体来说,利用相关蛋白质序列数据库和突变的结构上下文信息,生成一个0到1之间的连续分数,来近似评估突变的致病概率。
该连续分数允许用户根据自己的准确性要求,选择阈值将突变分类为致病性或良性。
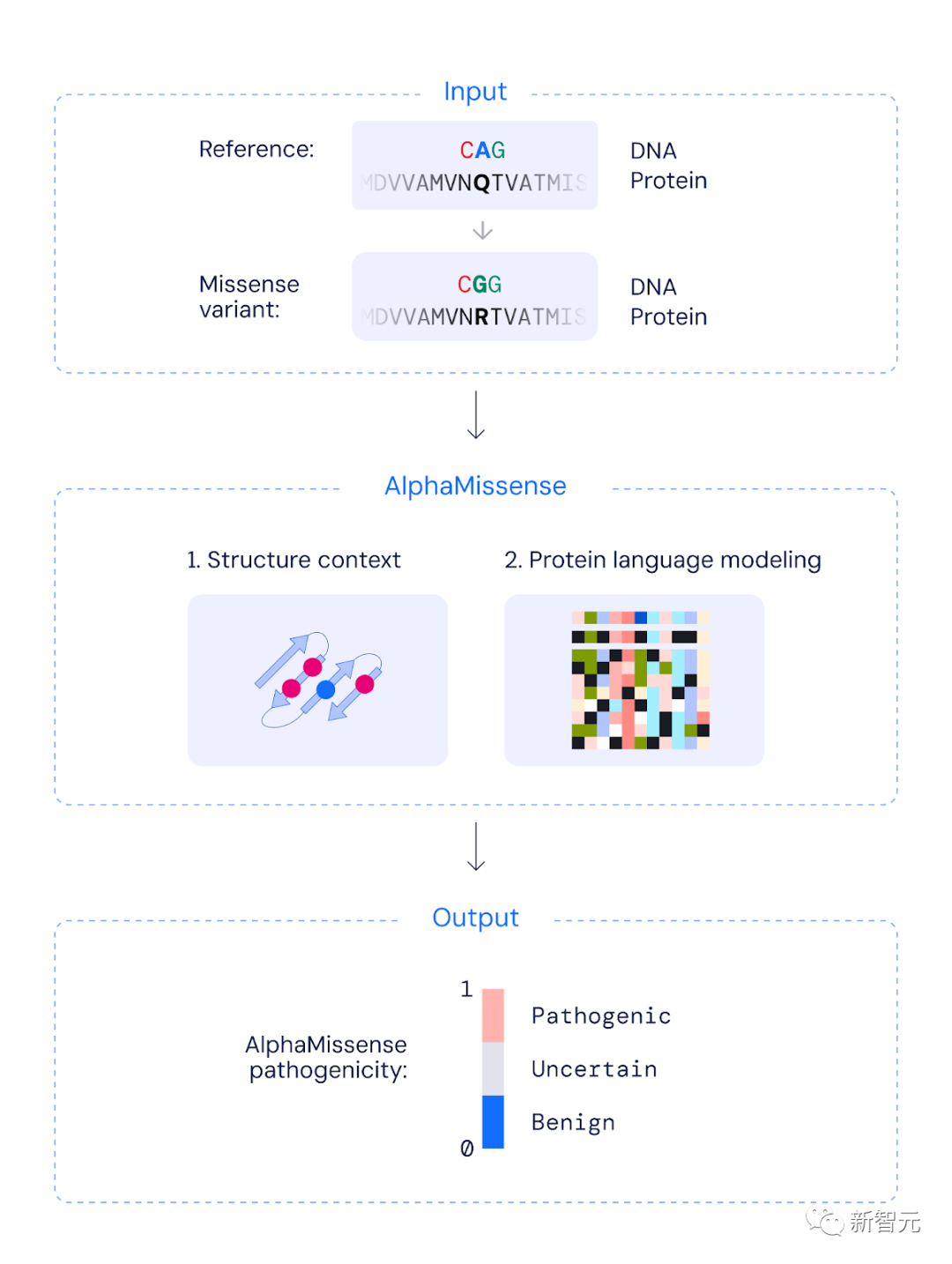
AlphaMissense如何对人类错义突变进行分类
在实验评估中,AlphaMissense在广泛的遗传和实验基准中实现了最先进的预测,而这一切都不需要对此类数据进行明确的训练。
在对来自ClinVar的变异进行分类时,AlphaMissense优于其他计算方法。ClinVar是一个关于人类变异与疾病关系的公共数据档案库。
AlphaMissense也是预测实验室结果最准确的方法,这表明它与衡量致病性的不同方法是一致的。
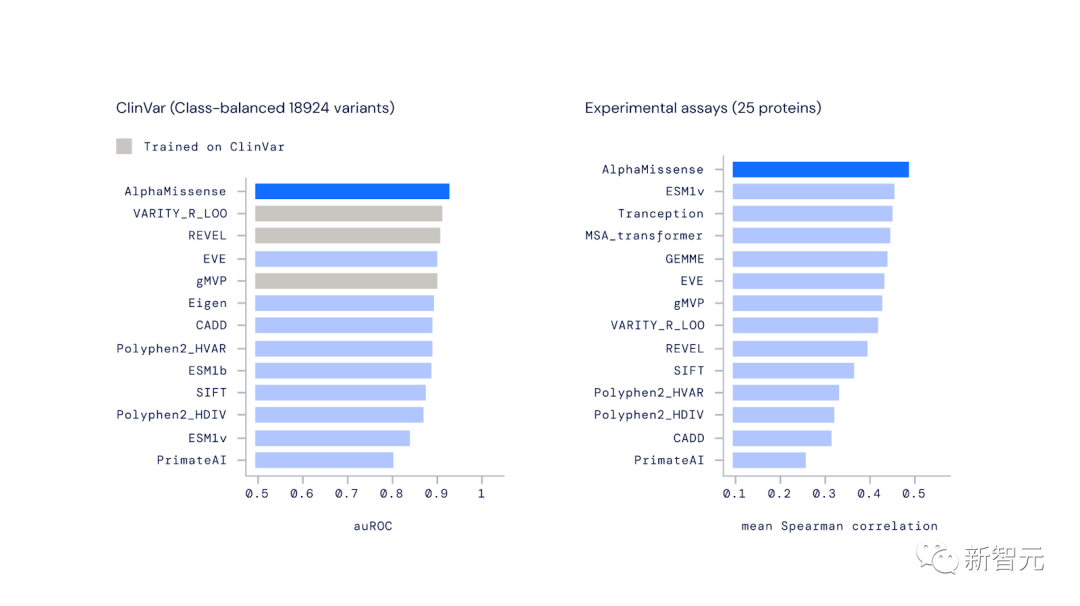
AlphaMissense在预测错义变体效应方面优于其他计算方法
这一举措帮助了全球数百万科学家加速研究,并为新的发现铺平了道路。
现在,以AlphaFold为基础的AlphaMissense,通过对DNA的溯源,进一步加深了全世界对蛋白质的了解。
同样的,转化这项研究的关键步骤是与科学界合作。
谷歌DeenpMind一直与英格兰基因组学组织合作,探索AlphaMissense的预测如何帮助研究罕见病的遗传学。
英格兰基因组研究所将AlphaMissense的研究结果与之前汇总的已知人类突变致病性数据进行了交叉对比。
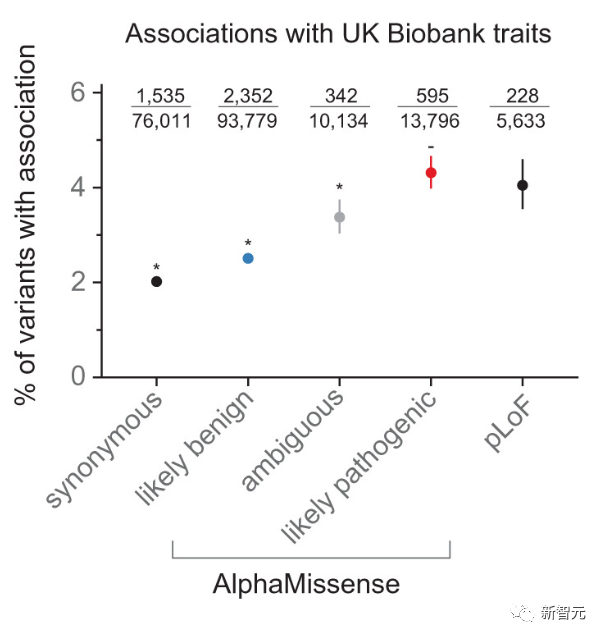
评估结果与AlphaMissense的预测一致,这为AlphaMissense提供真实世界的基准。
谷歌DeepMind公开了错义突变的查询表,并且分享了19,000多种人类蛋白质中所有可能的2.16亿个单氨基酸序列置换的扩展预测。
公开的数据中还包含了每个基因的平均预测值,类似于衡量一个基因的进化限制,表明该基因对生物体生存的重要性。
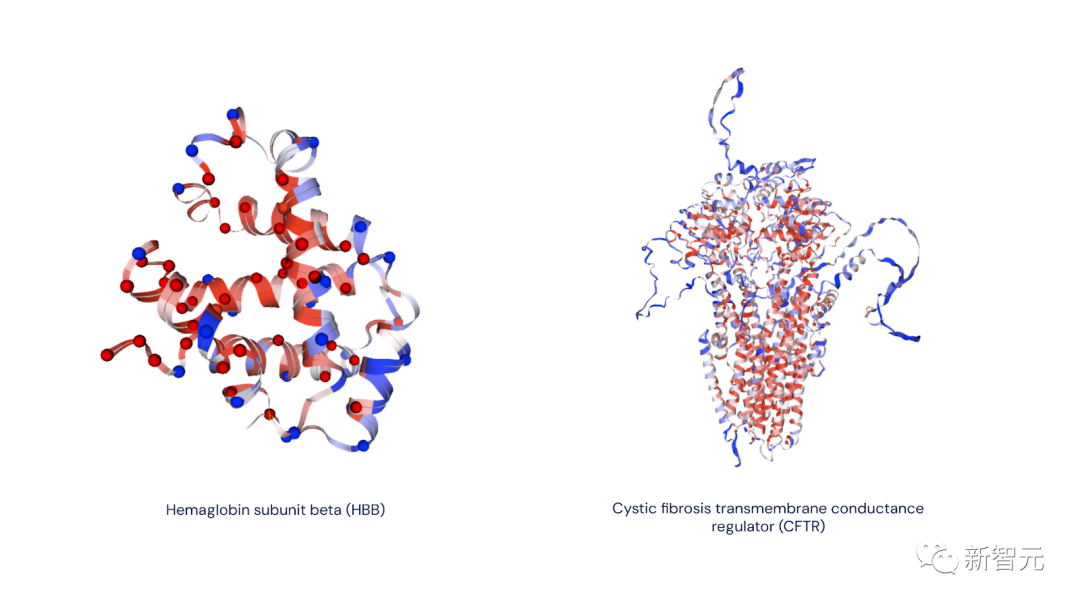
右图:囊性纤维化跨膜传导调节蛋白(CFTR 蛋白)。这种蛋白质的变异可导致囊性纤维化。
并且,谷歌DeepMind还与EMBL-EBI进行了合作。通过Ensembl突变效应预测器,研究人员将更方便地应用AlphaMissense的预测结果。
相信在不久的未来,AlphaMissense将帮助解决基因组学和整个生物科学的核心问题。


