
Science重磅:DeepMind再获突破,用AI开启理解电子相互作用之路

这一结果无疑会帮助科学家们更好地理解电子之间的相互作用,向着深入的研究进一步迈进,这也表明了深度学习(DL, Deep Learning)有望在量子力学水平上精确模拟物质——这可能使得研究人员在纳米水平上探索关于材料、药物和催化剂的问题,从而改进计算机的设计。简而言之,这是一次电子和分子的深度结合。
早在 50 多年前,这个用来描述量子物质基本性质的理论首次建立,实现了在量子水平上描述物质,该方法将电子在给定原子组中的位置与原子共享的总能量相关联,以确定分子的化学和物理特性。
作为人类智慧的结晶之一,很快地,它便成为物理、化学、材料科学等多个领域的强有力工具,是学习计算凝聚态物理/计算材料学/计算化学的必修基础理论。
通俗来说,密度指的是电子数密度,泛函则表示能量是电子密度的函数,而电子密度又是空间坐标的函数。那么,函数的函数,便称之为泛函(Functional)。也就是说,这是一种通过电子密度研究多电子体系电子结构的方法。
如果放到具体的操作中,DFT 可以通过各种各样的近似,把难以解决的包含电子-电子相互作用的问题化繁为简,变为无相互作用的问题,再将所有误差单独放进一项中(XC Potential),进而对误差进行分析。
然而,困扰已久的问题是:电子密度和相互作用能之间映射的确切性质,即所谓的密度泛函(density functional),仍然是未知的。
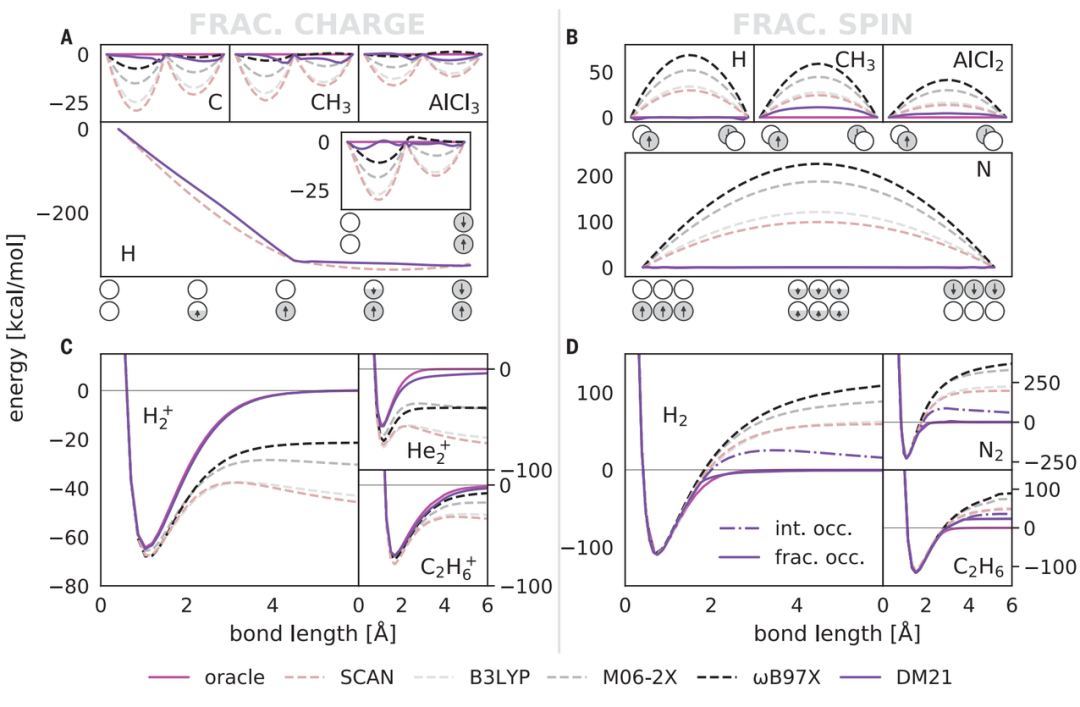
传统的 DFT 工具可以对具有一两个电子的系统进行建模,但它们无法对具有 1.5 个电子的系统进行建模,而这在一个电子被多个原子之间共享的情况下是很重要的。
一方面,这种带小数点的电子是虚构的物体,没有这样的电子,根据定义,电子是整体的,但是通过解决这些电子问题,我们能够正确描述化学系统。
正因为如此,即使是最先进的 DFT 在描述分数电子电荷(fractional electron charges)和自旋(spins)时,也会受到基本的系统误差的困扰。
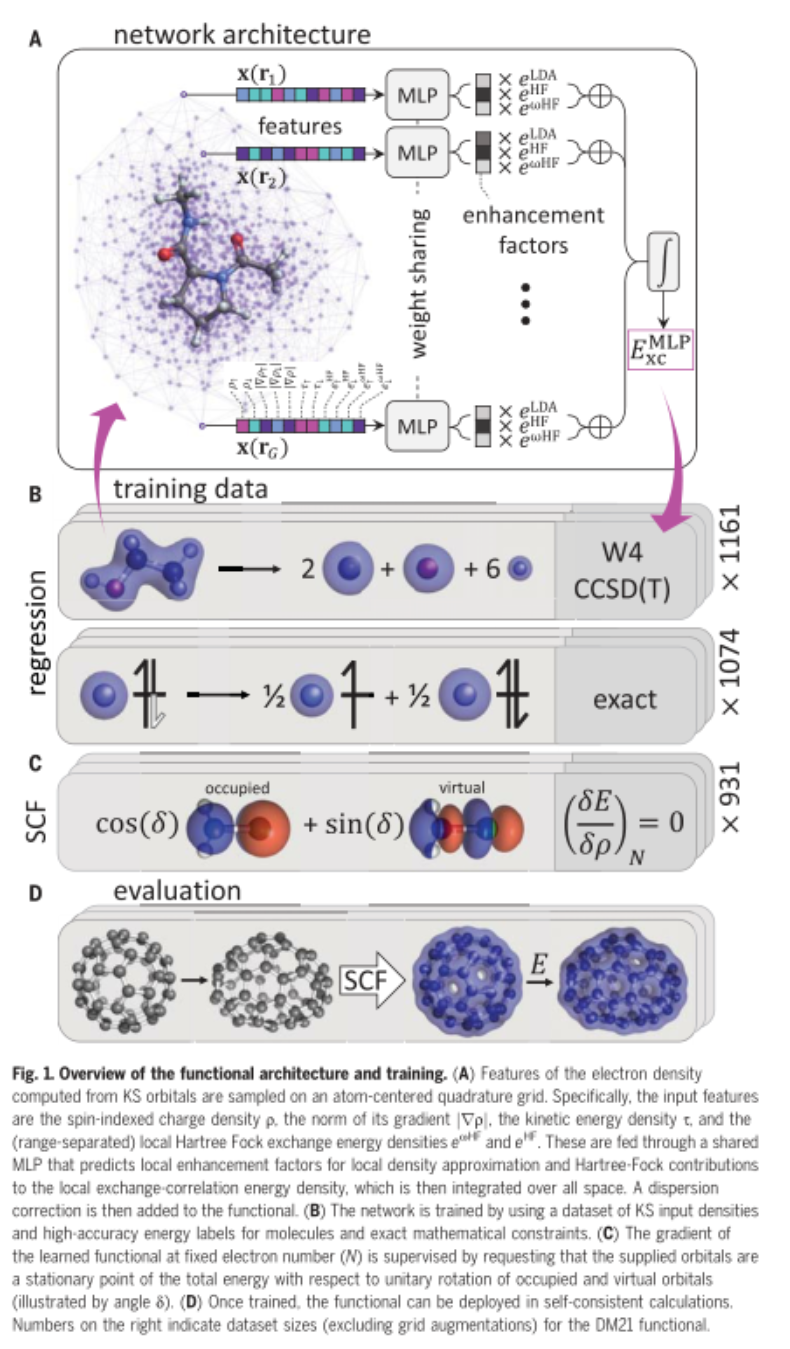
作为 DeepMind 的研究科学家,James Kirkpatrick 和他的同事使用 DeepMind 平台开发了“DM21”(DeepMind 2021)框架,可以利用精确的化学数据和分数电荷约束来训练神经网络。

研究人员用 2235 个化学反应示例训练了他们的人工智能,并提供了有关所涉及的电子和系统能量的信息。其中,1074 个代表了分数电子会对传统 DFT 分析造成问题的系统。
然后,他们将人工智能应用于未包含在训练数据中的化学反应。
DeepMind 21 不仅正确地表示了分数电子,而且其结果比传统的 DFT 分析更精确。它甚至可以处理关于具有奇怪属性的原子的数据,这些数据与训练数据中的任何东西都不相似。
DM21 正确描述了人工电荷离域和强相关性的典例,并且在主族(main-group)原子和分子的全面基准评估上优于传统泛函。此外,DM21 可以准确地模拟复杂系统,如氢键链(hydrogen chains)、带电荷 DNA 碱基对和双自由基体系的过渡态。
更为关键的是,该研究中的方案依赖于不断改进的数据和约束,因此,它代表着一条通向泛函的可行途径。
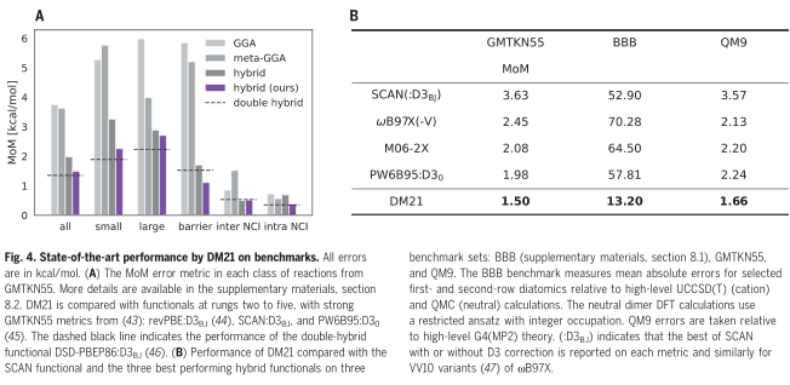
这个非常小的误差与大多数泛函误差相比,是由大量精心选择的成分和拟合的分子数据造成的。无论是否包含分数电荷和自旋数据,误差本质上是相同的。然而,这些数据的加入提高了 DM21 在电荷转移和强相关性问题上的性能。
Jon Perdew 在相关观点中写道:“由 Kirkpatrick 等人开发的 DM21 的重要性,并不在于产生了最终密度泛函,而是用一种 AI 方法解决了分数电子和自旋问题,该问题一直无法通过直接解析方法来创建泛函。”
整个研究工作表明,通过结合约束满足和 AI 拟合大而多样的数据集,可以设计出更具有预测性的精确密度函数。
DeepMind 演示了神经网络如何提高密度泛函的近似值,有力地显示了 DL 在量子力学水平上精确模拟物质的前景。此外,DeepMind 还开源了代码,为研究者提供了探索研究的基础。
对于这一成果,James Kirkpatrick 表示:“了解微观尺度现象对于帮助我们应对 21 世纪的一些重大挑战,从清洁电力到塑料污染,正变得越来越重要……这项研究朝着正确方向迈出的关键一步,使我们能够更好地理解电子之间的相互作用,而电子就是将分子粘在一起的‘胶水’。”
从短期来看,这将使研究人员能够通过代码的可用性,获得一个改进的精确密度函数的近似值;从长远来看,这是 DL 在量子力学水平上精确模拟物质的更进一步——这可能使研究人员能够在纳米水平上探索材料、药物和催化剂的问题,从而在计算机上实现材料设计。
评论