
NGS基础:测序原始数据批量下载
生物或医学中涉及高通量测序的论文,一般会将原始测序数据上传到公开的数据库,上传方式见测序文章数据上传找哪里;并在文章末尾标明数据存储位置和登录号,如 The data from this study was deposited in NCBI Sequence Read Archive under accession SRA: SRP114962.。
NCBI的SRA (Sequence Read Archive) 数据库(http://www.ncbi.nlm.nih.gov/sra/) 是最常用的存储测序数据的数据库。目前SRA数据的组织方式分为下面4个层次:
Studies—研究课题;
Experiments—实验设计;
Runs—测序结果集;
Samples—样品信息。
进入SRA官网:https://www.ncbi.nlm.nih.gov/sra, Search框中输入SRA编号(SRP114962),获得如下图的界面:
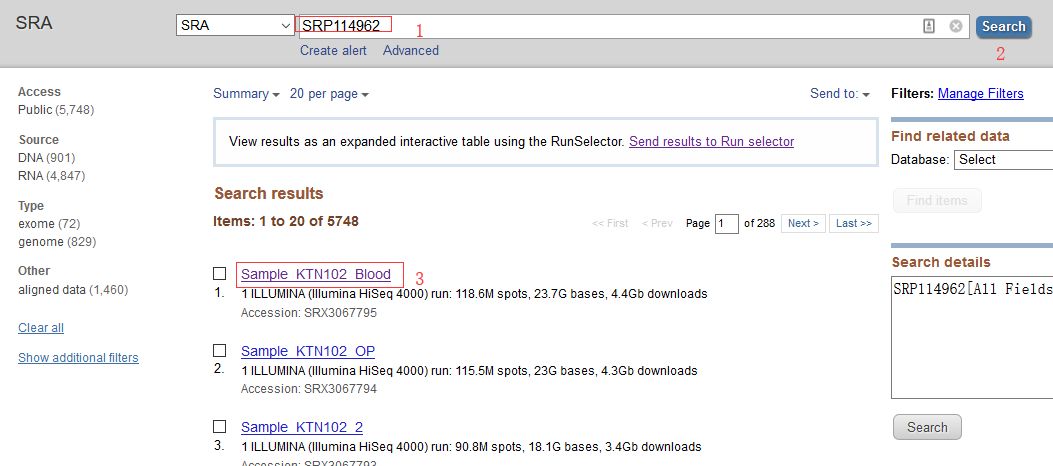
点击第一个样品即可查看其详细信息。

当样品比较多时,可以点击Send results to Run selector(图中画圈的位置)进入筛选页面。
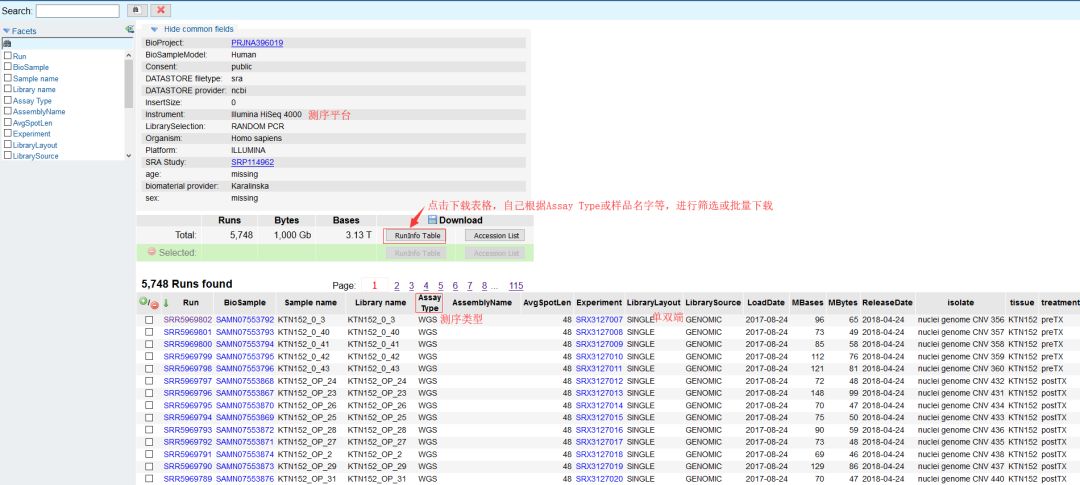
从图中可发现,测序平台是Illumina HiSeq 4000,5748个Runs,每个Run的名字、样本名、测序类型(全基因组/外显子组等)、tissue、treatment等。
在如此多的Runs中,假设我们想获取其中两个病人的化疗前和化疗后的外显子组测序数据,观察其化疗前后究竟有哪些基因突变以及突变的频率怎么样。数据来自于文章 肿瘤化疗无效是对预先存在的突变的选择还是诱发新突变,Cell给你答案。
5748个Runs,有116Page,怎么找呢?

在Facets下拉框中先勾选Assay Type,等待页面相应后勾选wxs,即全外显子组数据,等待页面相应。
在Facets下拉框中勾选Sample name,等待页面相应后勾选ktn102及ktn134两个病人的分别四个样本(四种treatment:pre、2cycleschemo、operative和blood),如图。等待页面相应。获得Run编号(蓝色框):SRR5908363、SRR5908362…
然后使用NCBI提供的工具SRAToolkit下载。
SRA toolkit https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software, 根据服务器操作系统类型下载对应的二进制编码包,下载解压放到环境变量即可使用。
使用NCBI提供的SRA-toolkit中的工具fastq-dump直接下载SRR文件,并转换为FASTQ格式,--split-3参数表示如果是双端测序就自动拆分,如果是单端不受影响。--gzip转换fastq为压缩文件,节省空间。
下载的数据集一般比较大,放入后台不中断下载 (nohup cmd &)。
nohup fastq-dump -v --split-3 --gzip SRR5908360 &
nohup fastq-dump -v --split-3 --gzip SRR5908361 &
nohup fastq-dump -v --split-3 --gzip SRR5908362 &
nohup fastq-dump -v --split-3 --gzip SRR5908363 &
nohup fastq-dump -v --split-3 --gzip SRR5906250 &
nohup fastq-dump -v --split-3 --gzip SRR5906251 &
nohup fastq-dump -v --split-3 --gzip SRR5906252 &
nohup fastq-dump -v --split-3 --gzip SRR5906253 &注意:如果数据量很大可能需要下载1-2天。数据下载完会在~/ncbi下面存在缓存的sra文件,记得定时清空。
按照上述步骤下载完毕后可看到很多个fastq.gz格式测序文件。
数据比较多时,一个个手动写也比较麻烦?怎么处理呢?

下载上面的metadata后自己生成一个批量下载并重命名的脚本。下下来的metadat文件通常名字是SraRunTable.txt,列很多,且是CSV文件,分隔符是逗号,某一个字段里面还有逗号的存在。Linux命令直接不好处理。

先写一个单行R脚本提取Run和Sample Name列,并另存到文件SraRunTable.tsv。
Rscript -e 'write.table(read.table("SraRunTable.txt",sep=",", header=T, row.names=NULL)[,c("Run","Sample.Name")],"SraRunTable.tsv",sep="\t", quote=F,col.names=T, row.names=F)'然后用awk就可以读取第一列批量下载并结合第二列批量重命名就可以了。
awk 'FNR>1{system("fastq-dump -v --split-3 --gzip "$1"; rename "$1" "$2" "$1"*");}' SraRunTable.tsv
等同于手动输入了如下命令
fastq-dump -v --split-3 --gzip SRR12603383; rename SRR12603383 PFER12d1 SRR12603383*
fastq-dump -v --split-3 --gzip SRR12603384; rename SRR12603384 PFER12d3 SRR12603384*
fastq-dump -v --split-3 --gzip SRR12603385; rename SRR12603385 PFER12d2 SRR12603385*
fastq-dump -v --split-3 --gzip SRR12603386; rename SRR12603386 PFER12d1 SRR12603386*
fastq-dump -v --split-3 --gzip SRR12603387; rename SRR12603387 PFER9d3 SRR12603387*
fastq-dump -v --split-3 --gzip SRR12603388; rename SRR12603388 PFER9d2 SRR12603388*
fastq-dump -v --split-3 --gzip SRR12603389; rename SRR12603389 PFER9d1 SRR12603389*
fastq-dump -v --split-3 --gzip SRR12603390; rename SRR12603390 PFER9d3 SRR12603390*往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集
(请备注姓名-学校/企业-职务等)