
男性不育致病基因的发现:过去,现在和未来

背景介绍
不育症是一种复杂的疾病,影响了全球约70%的男性人口,表现具有异质性,从先天性或后天性泌尿生殖器异常、内分泌失调、免疫因素到精子数量和质量的缺陷。除非借助辅助生殖技术,受影响的男性是无法自然传递其遗传信息的。未受影响的父母也可能通过将基因变异遗传给后代,从而导致后代发生常染色体隐性遗传或X连锁的男性不育。此外,正常配子发生期间也可能发生基因变异,这些新发变异也可能引起后代中的男性不育。
在大约15%的不育男性中,遗传缺陷可能是其产生这些疾病的根本原因。虽然核型分析、无精症因子(AZF)微缺失筛查以及囊性纤维跨膜转运调节物(CFTR)的变异分析等方法得到广泛使用,然而最近的一项大型队列研究表明,获得遗传学诊断的男性不育患者仅有4%。
对男性不育的遗传学认识始于二十世纪中叶,在不断更新的分子技术的帮助下一直持续到今天。尽管在20世纪50年代就因在Klinefelter综合征患者中发现额外的一条X染色体,而首次报道了男性不育的遗传学证据,但直到20世纪90年代末才加强了此方面的研究。最开始集中在雄激素受体(AR)和CFTR基因中的特异性缺失和变异以及Y染色体的异常研究。近年来,研究者使用了全基因组染色体微阵列以及下一代测序(NGS)等方法,将更有利于探索男性不育相关基因(图1)。与此同时,研究者、临床医生和患者加强合作以及信息共享,将会造福更多人。
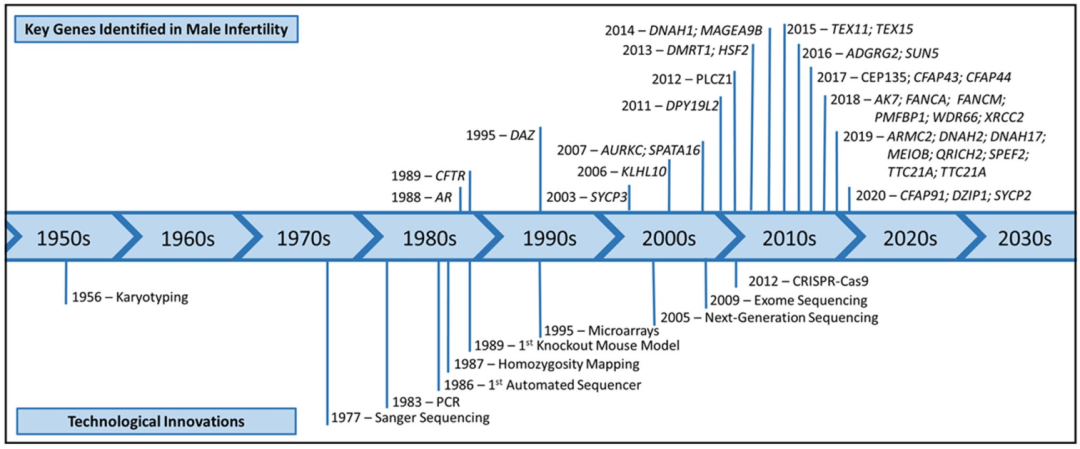 图1
图1
过去:染色体研究和特异基因分析提示了第一批男性不育基因
染色体异常导致男性不育
核型分析是第一个被用于分析不育男性是否存在遗传学异常的检测,直到今天仍然被广泛使用。这种细胞学遗传技术可以分析与男性不育相关的多种染色体异常,其中最常见的是Klinefelter综合征(47,XXY),存在于约15%的非阻塞性无精症患者中;核型分析与荧光原位杂交(FISH)技术相结合可以提示引起原发性不育和性发育障碍的其它染色体异常,特别是46,XX男性、罗伯逊易位和相互易位。
1976年在6例无精症患者中发现Y染色体q11远端的部分缺失,该区域被认为是精子形成所必需的。尽管这表明Y染色体缺失的区域存在控制人类精子发生的基因,但直到20世纪90年代才发现直接参与精子生成失败的第一个基因。研究者们通过聚合酶链式反应(PCR)和Y染色体特异序列标记位点(STSs)分析等方法来鉴定Y染色体缺失区域的候选基因。其中,无精子症缺失基因(Deleted in Azoospermia,DAZ)是最强的候选基因,目前已鉴定4个DAZ同源基因(DAZ1-4)。这些基于PCR的技术的使用进一步揭示了在精子生成失败男性中Y染色体上经常发生缺失的基因组区域,AZF区域a、b和c。每个区域中又包含在睾丸中高度表达或特异表达的精子发生相关的候选基因,包括BPY2、CDY、DAZ、HSFY、RBMY、PRY、TSPY、VCY和XKRY等。AZF区域的微缺失可引起2-10%的不育男性表现为从少精到无精的可变表型。
男性不育特异性基因变异的研究
1988年在X染色体上鉴定出第一个Y染色体以外的与男性不育相关的基因。当时,通过连锁分析定位到X染色体的鼠雄激素受体基因的变异可明确导致小鼠睾丸女性化。在这个基础上,Brown等人使用Southern印迹等方法揭示了人雄激素受体(AR,也称为NR3C4)的缺失导致轻度或部分雄激素不敏感综合征患者的不孕,以及完全雄激素不敏感综合征患者的性逆转。目前已在2%的不育男性中发现了可导致轻度或部分雄激素不敏感综合征的AR基因突变。1989年,使用限制性片段长度多态性(RFLP)分析等方法,7号染色体上CFTR基因的突变被发现是囊性纤维化的基础。自报道以来,研究表明该基因的特定突变会引起先天性双侧输精管缺如(CAVD)从而导致孤立性不孕。CFTR突变的携带者在全球人群中较高,然而CAVD是一种非常罕见的疾病,仅在1-2%的不育男性中发现。
Sanger测序在21世纪初被广泛应用于人类疾病研究。SYCP3基因突变的鉴定就是一个很好的例子,它与精子发生过程中减数分裂停止相关。先是在小鼠中确定了纯合SYCP3基因突变体由于精子发生期间的大量凋亡细胞丢失而不育,随后分离出人SYCP3基因后,发现具有相同的功能。## 现状:技术进步使不育基因组研究可进行无偏倚的分析
利用微卫星扫描和SNP微阵列筛选基因多态性
1990年代对现有测序技术的调整以及单核苷酸多态性(SNP)微阵列的引入使得男性不育基因组研究方法发生转变。最初,这项工作的重点是开发和应用大量的多态性标记来筛选近亲血统不育男性基因组中的纯合性区域。2007年,使用这种新的定位克隆方法鉴定出两个引起多种精子形态异常的男性不育新基因,`AURKC和SPATA16。也在不育男性队列中鉴定出DNAH1基因纯合变异可能导致精子鞭毛形态异常。
基于染色体微阵列的基因组拷贝数检测
SNP芯片技术的应用在2011年发现了一个与男性不育相关的重要CNV,即DPY19L2基因的200 kb纯合缺失。最初,DPY19L2基因的特异性扩增和测序受到假基因干扰。2012年,Coutton等人优化了条件得以特异性扩增和测序该基因,并鉴定出圆头精子症患者存在缺失突变和无义突变或错义突变组成的复合杂合突变,或者纯合错义突变。现在,已知DPY19L2基因的缺失突变及点突变最为常见。
2011年,Tüttelmann等人使用染色体微阵列技术在三个无精或少精患者中发现罕见的性染色体CNV变异。随后,Yatsenko等人鉴定出同样地X染色体上TEX11基因3个外显子缺失。TEX11基因的截短和剪接突变等致病性突变也相继被报道。运用相同方法,位于9号染色体上DMRT1基因的缺失也被报道跟无精子症相关。然而不仅是DMRT1基因的缺失,许多仅在少量甚至单个不育男性中报道的罕见CNV,其在不育中的功能仍然未知。NGS技术可以检测CNVs与SNVs(单核苷酸变异)等变异形式。最近,在一名严重少精子症患者中报道了一种影响SYCP2基因的平衡相互易位,最初是通过核型分析发现,但是进一步使用染色体微阵列和NGS进行明确变异。有趣的是,通过国际协作,发现SYCP2基因的致病性功能缺失突变也影响了德国的三位不育患者。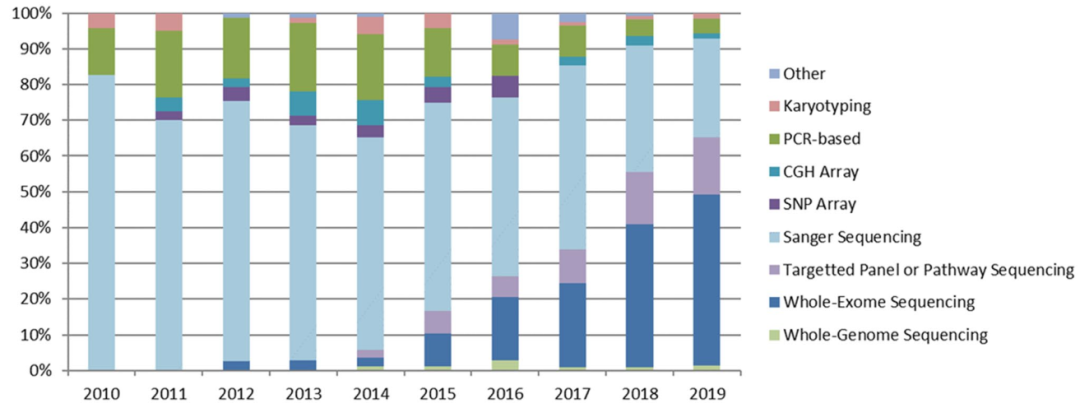 图2
图2
NGS用于新致病基因的检测
图2显示了近年来外显子组测序已成为男性不育症基因研究的主要技术。在精子鞭毛形态异常等方面,已发现许多重要的导致隐性疾病基因。同时,已发现睾丸特异性基因BRDT、SUN5和PMFBP1的纯合或复合杂合突变将破坏精子的头-鞭毛连接。由于较高的遗传异质性导致了的表型多样性,从生殖细胞完全缺乏(唯支持细胞综合征)到各种形式的成熟停滞,常见形式的非阻塞性无精子症(non-obstructive azoospermia,NOA)的遗传学机制研究较为困难。在这方面已经取得了重大进展,其中无精子症男性的外显子组测序在近亲和非近亲家族中提示了TEX15、FANCM、XRCC2、MEIOB、FANCA、ADGRG2等基因的致病突变。然而,许多这些新的候选基因尚待确定。
候选不育基因的临床验证
对于男性不育症的基因-疾病关系的确定,应考虑各种水平的证据,应继续补充和定期重新评估所有相关基因的遗传学和功能证据。同时,根据最新的技术发展设计正确的功能验证实验至关重要(图3)。
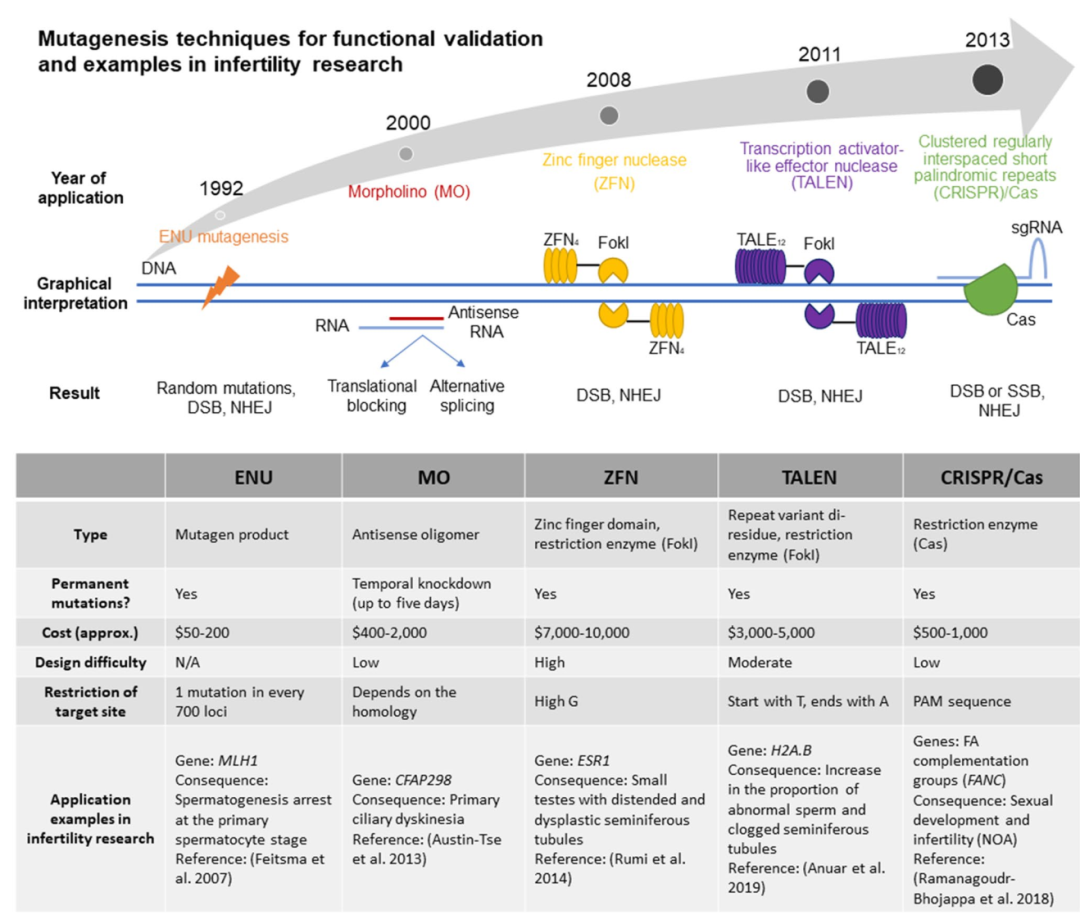 图3
图3
未来:克服现有的局限性以提高生物学理解,患者诊断和预后
无偏倚的NGS方法的引入彻底改变了许多疾病的遗传原因识别。尽管这些方法在研究实验室中经常用于研究男性不育症的遗传学,但它们尚未鉴定出引起该疾病的大多数基因。对于DFS-MMAF,无头精子综合症和小精子症等几种男性不育症,目前基于已知遗传原因的诊断率约为50%。相比之下,男性不育的最常见形式即无精子症和少精子症的诊断率仍然非常有限。
发现相对常见的异质性疾病的致病基因中的挑战
外显子组,尤其是全基因组测序,使我们能够进行无偏倚的遗传学研究,从而有助于鉴定新的疾病基因或基因组区域。然而,谨慎的表型和队列选择至关重要。这种情况下,大型患者和对照队列以及可靠的统计方法对于鉴定患者基因组中突变负荷增加的基因至关重要。这要求进行大规模的协作研究和广泛的数据共享,以可靠,可靠地识别和功能验证新的男性不育基因。最近在雄性不育遗传学领域建立了国际联盟以促进这一发展,包括GEMINI联盟(https://gemini.conradlab.org/)和`IMIGC`联盟(https://www.imigc.org/)。
建立男性不育症中遗传模式和新发突变研究
在男性不育研究中应用的主要挑战之一是通常无法获得父母的DNA样本。然而,最近几个小组已经开始为男性不育症基因研究组建患者-父母三人检测模式,旨在鉴定引起男性不育症的新发突变,并增加对亲本遗传模式的理解。
全面理解男性不育症的所有基因组变异
大多数NGS方法使用短读测序,这限制了重复结构、高度同源序列和结构变异的检测。考虑到Y染色体高度同源和重复区域中染色体微缺失的重要性以及其他基因组区域中的结构变异,这对男性不育研究产生了重大影响。最近,出现了新的测序平台,其测序读取长度从几百个核苷酸增加到了10kb,甚至更长。由于它们的读取长度增加,因此这些平台能够更好地检测重复扩增,同源序列和结构基因组变异。不幸的是,这些技术的当前碱基读取准确性和成本尚待提高。然而,在未来十年内,将可能获得完整、准确且负担得起的人类基因组测序,男性不育的主要挑战将变成变异解释,而不是检测。

往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集
(请备注姓名-学校/企业-职务等)