RNA-seq最强综述名词解释&思维导图|关于RNA-seq,你想知道的都在...
前言
NGS系列文章包括NGS基础、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述))、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step) - Limma差异分析、火山图、功能富集)等内容。

之前整理的一篇大综述 — Nature重磅综述 |关于RNA-seq,你想知道的都在这收到了热烈反响,阅读人数过万。
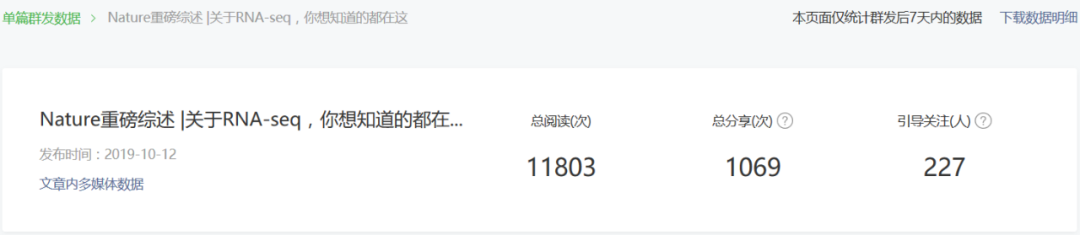
行文很长,最后精炼下来的文字近三万,适合深度阅读思考。
上次发出时,有读者留言说部分专业名词不理解。为了方便理解和对综述有个概览,特整理了下面的思维导图,对应原文,共计8个大标题,大标题下又分有小主题,各个分支介绍有每个主题的主要内容及采用方法。
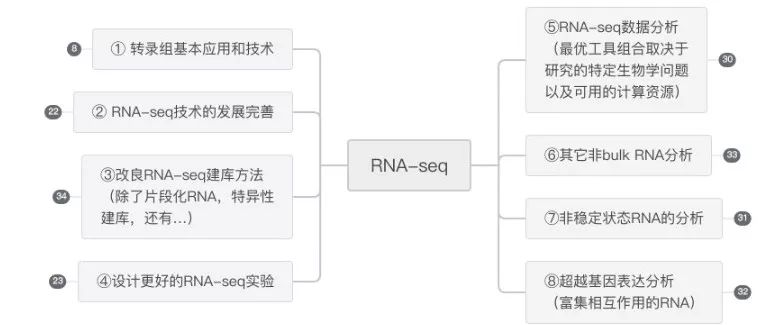
内容已发布在石墨文档,链接如下:
https://shimo.im/mindmaps/qQVV3r3Pqx8DVGjC/ 《RNA-seq思路图(欢迎大家备注、修改,可先创建副本,在副本文件修改)》,可复制链接后用石墨文档 App 或小程序打开
Note:想要打开全部分支、添加备注或修改信息,请先创建副本,在备份文件打开修改,原文件不支持修改
原文在深度总结了RNA-seq这些年的同时,还分享了文中一些名词的解释,编译分享如下,希望有助于进一步理解学习。
Read depthRead深度:一个样本测序得到的reads数;容易和基因组测序的覆盖度 (多少基因组区域被测到了)和测序深度混淆 (单个核苷酸被测到的次数或所有核苷酸被测到的平均深度)。Short-read短读长:测序得到的长度最大是500 bp的reads,常见的测序片段长度为100-300 bp;本文中的短读长测序片段代表测到的mRNA片段和降解了的mRNA。Long-read长读长:测序得到的超过1000 bp的reads,本文中代表全长或近乎全长的mRNA。Direct RNA sequencing(dRNA-seq): 直接测序RNA而非cDNA的测序技术,通常用于测序全长或近全长的mRNA 。Multi-mapped reads多重比对的reads:从转录组同源区域测序得到的reads,不能精确确认其转录本或基因组的来源。Synthetic long reads合成long reads:通过组装多个短读长得到长读长的方法。唯一分子标识符(
UMIs):在扩增前,构建RNA-seq文库的时候加入的短序列或barcodes,理想情况下每条转录本结合一个唯一的标识符,含有此标识符的reads都来源于此转录本,定量时只计算一次。可以用来降低RNA-seq的定量偏好性,在RNA起始量低的单细胞实验中尤为适用。Read length读长:单个测序reads的长度,short-read RNA测序得到的长度通常是50-150 bp。Sensitivity敏感性:样本中多大比例的转录本会被测到,敏感性越高,这一比例越高。它受样本处理、文库制备、测序和计算偏好性的影响。Specificity特异性:度量差异表达转录本被正确鉴定出的比例的方法,它受样本处理,文库制备,测序和计算偏好性的影响。Duplication rates重复Reads比率:比对到转录组相同位置的的测序reads的比例。在RNA-seq文库中,一些转录本可能有高的重复率,因为它们在样本中表达水平高。高表达的基因的重复率很高,而低表达基因的或许有着最小的重复率。由此RNA-seq面临着一个挑战,该技术中大部分重复可能是高表达转录本带来的真实信号,而另一些则是由于扩增和测序偏好性造成的。Single-end sequencing单端测序 (SE):只测序cDNA片段的一端,因其费用低,常用于只关注差异基因表达的项目中。(NGS基础 - 高通量测序原理)Paired-end sequencing双端测序 (PE):cDNA片段两端分别测序,可以测序到cDNA的更多碱基,更好的识别剪接位点,常于差异基因表达分析项目。生物学重复:对生物来源不同的样本的多次检测,比如来自三个个体的组织,用于捕获生物个体自身的变化;这个变化要么是待研究的对象,要么是噪音。相较之下,技术重复是对同样的样本做重复的操作—比如,对一个组织做三次处理。
Expression matrix表达矩阵:差异表达RNA-seq项目的核心数据文件。每一行代表一个RNA,比如基因或者转录本。每一列是一个测序的样本。矩阵中的数值是每个RNA的reads数。这些可能是对转录异构体的计数估计,并通常在后续的分析前先进行标准化转化。Spike-in control内参:按特定浓度添加到样品中的外源核酸库。它们通常是预先合成的不同浓度的RNA,用于监测反应效率和技术方法的偏差和假阴性结果。Spatialomics空间转录组学:能保留给定样本(通常是组织切片)中每个转录本的空间信息的转录组分析方法。Nascent RNA新生RNA:刚刚转录出来的RNA,与已经加工并运输到细胞质的RNA相对应。Translatome翻译组:细胞、组织或生物体中正在翻译成蛋白质的mRNA集合。Structurome结构组:细胞、组织或生物体中RNA的二级和三级结构集合。Interactome互作组:细胞、组织和生物体中分子相互作用的集合,包括有RNA-RNA或者RNA-蛋白质的相互作用。Differential gene expression (DGE)差异基因:两个实验组中表达显著变化的基因。
你可能还想看
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集