
【数据竞赛】天池-蛋白质结构预测大赛冠军方案分享
1. 赛题介绍
赛题链接:https://tianchi.aliyun.com/competition/entrance/231781/introduction
本题为根据蛋白质的一级结构预测其二级结构,经过比赛期间组内师兄的讲解,我对蛋白质一级结构二级结构的理解如下,如有错误,欢迎指正。
蛋白质可以看成是一条氨基酸序列,在空间中是一种相互交错螺旋的结构,像一条互相缠绕的绳子:
这种三维结构叫做蛋白质的三级结构,而如果不考虑结构的三维性,或者说把这整条序列拉直,用一个一维的序列表示,这便是得到了蛋白质的一级结构:
GPTGTGESKCPLMVKVLDAV······
这些字母G、A、V等便是代表一个个的氨基酸,其中主要包含有20种常见的氨基酸。
用这样的序列表示蛋白质比起原始的三维结构确实方便不少,但却丢失了三维的结构信息,蛋白质的结构决定其功能,这里的结构不止是序列本身,更多的还依赖其三维结构。因此,便出现了蛋白质的二级结构,它是一条与一级结构长度相等的一维序列,用以表征一级结构种的各位置的氨基酸在三维空间种的形态,以保留一部分的三维结构信息,例如以上蛋白质一节结构对应的二级结构为:
EEEEEEETT······
==注意:以上二级结构最前面有11个空白符,这个空白符也是在蛋白质在三维空间种的一种松散结构表示。==
这里的' '、'E'、'T'等都是对应位置的氨基酸在空间种的形态(与一级结构 GPTGTGESKCPLMVKVLDAV······ 是一一对应的),例如'T'代表的就是该位置的氨基酸在空间中是一种氢键转折的形态。
本赛题就是需要通过蛋白质的一级结构,预测其二级结构,在深度学习种是一种典型的N-N的seq2seq问题。
2. 赛题理解
不难想到,蛋白质三维结构的形成,其实主要是受某些力的作用,不同氨基酸的分子量、体积、质量等性质都有差异,这些小分子间会受到分子间作用力的影响,换句话说,分子间作用力等多种因素共同作用,让蛋白质形成了这样的一种相对稳定的空间结构,以达到一种稳态;而倘若你强行把它拉直,它也会由于受力不均,又开始相互缠绕,以达到稳态。
因此,对于某条蛋白质的二级结构中第i个位置的空间形态,其不止是取决于对应一级结构中位置i的氨基酸,还取决于位置i周围氨基酸甚至整条序列的情况。
定义一级结构中位置i及其上下文的整条片段为X,对应的二级结构中位置i的形态为Y,我统计了整个训练数据中 P(Y|X) 的情况,并计算了在不同窗口大小时,P(Y|X)>0.95 在所有 P(Y|X) 中的占比情况如下表:
| 窗口大小 | 1 | 3 | 5 | 7 | 9 | 13 | … |
|---|---|---|---|---|---|---|---|
| P(P(Y|X)>0.95) | 0.00588 | 0.02188 | 0.55728 | 0.83511 | 0.84413 | 0.85431 | … |
以上结果也验证了之前的理解,且不难看出,当窗口大小达到7以上时,可以达到较好的预测。
3. 思路分享
这类题首先需要解决的是输入序列的编码问题,很自然的可以想到onehot和word2vec两种编码方法,本次赛题我们都进行了尝试。
3.1 Onehot与基本理化性质编码+滑窗法+浅层NN
氨基酸的基本理化性质包括分子量、等电点、解离常数、范德华半径、水中溶解度、侧脸疏水性,以及形成α螺旋可能性、形成β螺旋可能性、转向概率等(来自Chou-Fasman算法),这些数据百度都很容易找到。
然后是窗口大小的选择。经过测试,隐层节点数为1024,当窗口大小达到79以上时,线下MaF达到饱和,为0.749。再调节隐层大小为2048,最后的线下MaF为0.767。
==(注:此处为氨基酸级别的MaF得分,非官方评分方法的序列级MaF再平均的结果;且此处未进行交叉验证,仅仅是单模的结果,后面的线上结果也是,所以可能会有一定偏差)。==
该模型提交后线上结果为0.7312。(滑窗模型其实等价于基于整条序列的CNN模型)
3.2 Word2vec+深层NN
NN的结构设计主要参考论文《Protein Secondary Structure Prediction Using Cascaded Convolutional and Recurrent Neural Networks》[1],这是一篇使用深度学习进行蛋白质二级结构预测的经典论文,文中使用了CNN+BiGRU的结构进行蛋白质二级结构预测,模型结构如下:
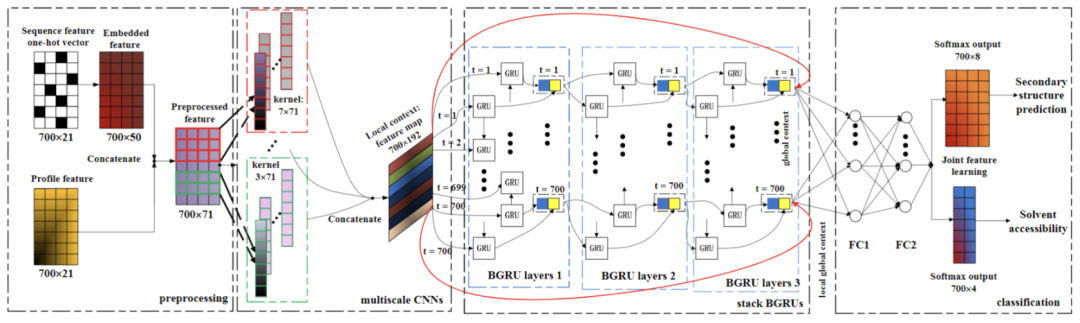
该模型先通过CNN捕获局部信息,再通过RNN融入全局信息,是NLP长文本任务的常见baseline模型。这里基本照搬了模型结构,但将编码部分改为了word2vec预训练的结构,词向量大小为128,其它结构和参数与原文一致,文章可从github项目目录进行下载。
此处还需注意一定的是,如果embedding层是单独对每个氨基酸进行编码的话,那么词表大小为23(数据集中共23种字母)。而在NLP种经常用到的一种叫做n-gram的技术,即将多个词绑定在以此形成整体,这个技术在蛋白质序列种也用得比较多,成为k-mers。倘若使用k-mers构建词表的话,假定k=3,那么词表的大小就是232323=12167,这样相当于在编码时将上下文也考虑了进去,增加了词的多样性,在一定程度上可以提高模型的学习能力,但也会增大过拟合的风险。
这里我也分别尝试了k=1和k=3的两个模型情况,线下分别为0.719和0.706,线上分别为0.7576和0.7518。
==(注:此处计算线下分数的方法与官方提供的是一致的,但不知道为何线下比线上低了这么多,暂时还未找到原因;且此处未进行交叉验证,仅仅是单模的结果,后面的线上结果也是,所以可能会有一定偏差)==
这两个模型的输入和数据划分都有较大差异,显然会有一定的融合收益,将二者的结果进行加权平均后,线上结果为0.7702。
3.3 最终模型
将以上几个模型的特征输入都有着较大不同,进行简单加权融合后,线上结果到达0.7770。
在得到以上结果后,进一步分析问题:
1.模型真正学会的到底是什么: 我们结合赛题理解部分的统计结果,不难想到,与其说模型是学会了推理,不如说模型主要是记忆了大量由X->Y的固定映射或搭配,然后根据不同搭配的置信度进行决策,学会如何权衡不同搭配以得到更加正确的结果,这也是选用单层小窗口CNN时严重欠拟合的原因。
2.氨基酸的编码表示: 在NLP任务中的字词编码由于词表过大导致维度过多且稀疏,这才出现了word2vec算法以得到词语的低维稠密表示,且不同字词间有很大的联系,而这一联系可以通过词向量间的cos距离等来刻画。而蛋白质总共包括的氨基酸种类较少,在本数据中只有23种,只需要一个23维的onehot向量就可以表示,且不同氨基酸间的关联度很小,更多的是差异性,而onehot向量是可以充分表达这一差异性的(不同onehot向量在高维空间中相互垂直)。这也是简单的onehot编码+大窗口CNN能如此有效的原因,也验证了前面的观点,即模型主要是记忆了大量由X->Y的隐射,预测时根据所记忆的大量先验知识,对输出进行决策。
综上,我们设计了最终的模型:在3.2部分的模型中,将embedding部分改成了25维onehot编码+14维理化特征+25维word2vec特征,其中onehot和理化特征部分在训练过程中是frozen的,而word2vec会随着训练进行finetune;其次是加大了CNN部分的窗口,设置成了[1,9,81]。
PS:这部分没有尝试其它的数值,这里是玄学设计,取了一个单粒度的窗口1(相当于最普通的神经网络,仅仅是对特征进行了非线性变换);大窗口81(为了达到之前的最优窗口79);以及大窗口开根号的数值——9,以折个中)
最终按次方案训练了一个3折的模型,线下MaF平均为0.756。
==(注:此处计算线下分数的方法是padding后序列级别的MaF再平均,理论上应该会高于去掉padding后的结果,这里同样线上的结果也好于线下==
将3折的模型加权平均后线上分数为0.7832。(榜上最优结果0.7855是融合了之前的几个模型,但没多大参考价值,最终模型可以说是融合了之前的所有模型,融合没多大价值,收益基本来自数据的分布差异)
4. 代码开源
代码在github进行了开源,基于pytorch,其中主要包含:
nnLayer:基本神经网络结构的封装。DL_ClassifierModel:整个模型的封装,包含训练、模型的加载保存等部分。utils:数据接口部分的封装。metrics:评价指标函数的封装。SecondStructurePredictor:模型的预测接口类。
使用方法如下:
# 导入相关类
from utils import *
from DL_ClassifierModel import *
from SecondStructurePredictor import *
# 初始化数据类
dataClass = DataClass('data_seq_train.txt', 'data_sec_train.txt', k=1, validSize=0.3, minCount=0)
# 词向量预训练
dataClass.vectorize(method='char2vec', feaSize=25, sg=1)
# onehot+理化特征获取
dataClass.vectorize(method='feaEmbedding')
# 初始化模型对象
model = FinalModel(classNum=dataClass.classNum, embedding=dataClass.vector['embedding'], feaEmbedding=dataClass.vector['feaEmbedding'],
useFocalLoss=True, device=torch.device('cuda'))
# 开始训练
model.cv_train( dataClass, trainSize=64, batchSize=64, epoch=1000, stopRounds=100, earlyStop=30, saveRounds=1,
savePath='model/FinalModel', lr=3e-4, augmentation=0.1, kFold=3)
# 预测, 得到的输出是一个N × L × C的矩阵,N为样例数,L为序列最大长度,C为类别数,即得到的是各序列各位置得到各类别上的概率。
model = Predictor_final('model/FinalModelxxx.pkl', device='xxx', map_location='xxx')
model.predict('seqData.txt', batchSize=128)
参考文献
[1]Li Z, Yu Y. Protein secondary structure prediction using cascaded convolutional and recurrent neural networks[J]. arXiv preprint arXiv:1604.07176, 2016.
往期精彩回顾
获取本站知识星球优惠券,复制链接直接打开:
https://t.zsxq.com/qFiUFMV
本站qq群704220115。
加入微信群请扫码: