
Cell 文章代码重改复现测试
有的人出生就在罗马

1引言
之前复现一篇 Cell 的文章,分析其代码有问题,按照文章的代码进行了分析:

后面基于比对到 转录组 的方法,重写了代码进行分析复现,但是代码都是基于转录组的:
我们看文章的话,文章是比对到 基因组上的, 软件用的也不是我们使用的 bowtie,而是 tophat 软件:

这次根据文章的方法使用 tophat 软件比对,然后对作者的代码进行重新修改。然后复现之前的结果。
2比对到基因组
这里的数据还是拿之前去除 rRNA 和 tRNA 序列的测序数据:
for i in SRR5188601 SRR5188602 SRR5188603 SRR5188604 SRR5188613 SRR5188614 SRR5188615 SRR5188616
do
tophat2 -p 20 -a 4 -i 40 -I 2000 \
-g 1 --max-insertion-length 0 \
--max-deletion-length 0 \
--no-novel-juncs \
--no-convert-bam \
-G index/Saccharomyces_cerevisiae.R64-1-1.105.gtf \
-o 5.map-data/${i} \
./index/genome-index/Saccharomyces_cerevisiae-genome \
4.rmrRNA-data/${i}.rmrRNA.fq.gz
done
比对率在八九十左右,质量还是非常高的。
3计算 density
代码的问题是从计算 density 开始的,所以从这步进行修改。
思路是加上每个染色体编号的标识来计算 density 就行了。
(julia代码编写)
# load package
using XAM
# define function
function CalculateCWRibosomeDensity(inputFile,outputFile)
# save in dict
density_dict = Dict{String,Float64}()
# open sam file
reader = open(SAM.Reader,inputFile)
record = SAM.Record()
# loop
while !eof(reader)
empty!(record)
read!(reader, record)
# do something
if SAM.ismapped(record)
# tags
refname = SAM.refname(record)
align_pos = SAM.position(record)
read_length = SAM.seqlength(record)
...
mkdir("./3.density-data")
# sample name
sample = ["Ssb1-trans-rep1","Ssb2-trans-rep1","Ssb1-inter-rep1","Ssb2-inter-rep1",
"Ssb1-trans-rep2","Ssb2-trans-rep2","Ssb1-inter-rep2","Ssb2-inter-rep2"]
SRR = ["SRR5188601","SRR5188602","SRR5188603","SRR5188604",
"SRR5188613","SRR5188614","SRR5188615","SRR5188616"]
# batch run
for i in range(1,8)
CalculateCWRibosomeDensity(SRR[i]*".sam","./3.density-data/"*sample[i]*".denisty.txt")
end
这里计算出来的结果有四列,分别是染色体,比对位置,raw density 和 RPM:
I 100217 0.1111111111111111 0.0030730133056879514
I 100218 0.1111111111111111 0.0030730133056879514
I 100219 0.1111111111111111 0.0030730133056879514
...
4填充间隔,滑窗分析
后面作者又用了一些代码 对 density 的其它基因组位置进行填充, 计算 RPM 和 滑窗平滑化,这里我直接一步到位。
思路是先对每条染色体的长度里的 density 进行填充,然后在全基因组水平进行滑窗平滑化。 (julia代码编写)这里需要提供 基因组数据 来获取每条染色体的长度信息,方便为填充数据和平滑化分析。
using FASTX
# save chromsome length
chromlenDict = Dict{String,Int64}()
# get length
open(FASTA.Reader, "../index/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa") do reader
record = FASTA.Record()
while !eof(reader)
read!(reader, record)
## Do something.
chromlenDict[FASTA.identifier(record)] = FASTA.seqlen(record)
end
end
Dict{String, Int64} with 17 entries:
"VIII" => 562643
"XVI" => 948066
"V" => 576874
"III" => 316620
"XIII" => 924431
"IV" => 1531933
"XII" => 1078177
"X" => 745751
"I" => 230218
"XI" => 666816
"XIV" => 784333
"XV" => 1091291
"Mito" => 85779
"VII" => 1090940
"II" => 813184
"IX" => 439888
"VI" => 270161
计算:
using FASTX
function slidewindowOnGenome(geonme,inputFile,outputFile)
################################################################
# 1.get genome chromosome length
################################################################
# save chromsome length
chromlenDict = Dict{String,Int64}()
# get length
open(FASTA.Reader, geonme) do reader
record = FASTA.Record()
while !eof(reader)
read!(reader, record)
## Do something.
chromlenDict[FASTA.identifier(record)] = FASTA.seqlen(record)
end
end
################################################################
# 2.load ribosome denisty file
################################################################
densityDict = Dict{String,Tuple}()
open(inputFile,"r") do denistyFile
for line in eachline(denistyFile)
chrom,alignPos,raw,rpm = split(line)
densityDict["$chrom|$alignPos"] = (parse(Float64,raw),parse(Float64,rpm))
end
end
...
# sample name
sample = ["Ssb1-trans-rep1","Ssb2-trans-rep1","Ssb1-inter-rep1","Ssb2-inter-rep1",
"Ssb1-trans-rep2","Ssb2-trans-rep2","Ssb1-inter-rep2","Ssb2-inter-rep2"]
# batch run
for i in range(1,8)
slidewindowOnGenome("../index/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa",
"./3.density-data/"*sample[i]*".denisty.txt",
"./3.density-data/"*sample[i]*".swfill.denisty.txt")
end
输出结果:
VIII 1 0 0 0.0 0.0
VIII 2 0 0 0.0 0.0
VIII 3 0 0 0.0 0.0
...
结果包含六列,分别为染色体,染色体位置,raw density,RPM,slided(raw density) 和 slided(RPM)。
5metagene analysis
首先我们需要准备基因信息文件(python代码编写):
# get protein-coding gene start and end info
geneList = open('gene-info.txt','w')
with open('Saccharomyces_cerevisiae.R64-1-1.105.gtf','r') as gtf:
for line in gtf:
if line.startswith('#'):
continue
fileds = line.split()
# for Plus strand
if fileds[2] == 'gene':
if len(fileds) > 14:
type = fileds[15]
gene_name = fileds[11].replace('"','').replace(';','')
else:
type = fileds[13]
gene_name = fileds[9].replace('"','').replace(';','')
if type == '"protein_coding";':
gene_name = gene_name
start = fileds[3]
end = fileds[4]
strand = fileds[6]
geneList.write('\t'.join([gene_name,str(start),str(end),strand,str(fileds[0])]) + '\n')
else:
pass
else:
pass
geneList.close()
结果如下:
SOR2 8683 9756 - IV
AAD4 17577 18566 - IV
CIN10 1248154 1249821 - IV
YDL094C 289572 290081 - IV
THI74 1338274 1339386 + IV
SPS1 1485566 1487038 - IV
分别为基因名,基因起始位置,基因终止位置,所在链,染色体编号。
因为 酵母基因基本上都是一整个 CDS 区域,UTR 区域和内含子区域基本上没有,所以可以像上面这样准备结果。对于人及小鼠等高等生物,得考虑 不同位置的 CDS 区域, UTR 区域, 不同异构体等信息,会复杂得多。代码就得重新编写。
计算:
def metageneAnalysis(inputFile,outputFile,geneinfo):
mylist = range(-50, 1501)
rangeDict = dict([i, 0] for i in mylist)
countDict = dict([i, 0] for i in mylist)
# load denisty file
DictDensity = {}
inFile1 = open(inputFile, 'r')
line = inFile1.readline()
while line != '':
fields = line.split()
chrom = fields[0]
pos = fields[1]
density = float(fields[3])
DictDensity["|".join([chrom,pos])] = density
line = inFile1.readline()
inFile1.close()
# Upload gene list
inFile2 = open(geneinfo, 'r')
line = inFile2.readline()
while line != '':
fields = line.split()
col0 = str(fields[0]) # gene name
col1 = int(fields[1]) # start of gene
col2 = int(fields[2]) # stop of gene
col3 = str(fields[3]) # strand of gene
col4 = str(fields[4]) # chromesome of gene
length = abs(col1 - col2) + 1
if col3 == '+':
# Select genes
if length > 400: # use genes longer than 400 nt; this value can be changed
normVal = 0
for Z in range(col1, col2 + 1):
key = "|".join([col4,str(Z)])
if key in DictDensity:
normVal += DictDensity[key] # determine expression level
if normVal > 50: # continue if sum of reads is greater than 50; this can be changed if desired
...
import os
os.mkdir('4.metagene-data')
# sample name
sample = ["Ssb1-trans-rep1","Ssb2-trans-rep1","Ssb1-inter-rep1","Ssb2-inter-rep1",
"Ssb1-trans-rep2","Ssb2-trans-rep2","Ssb1-inter-rep2","Ssb2-inter-rep2"]
# batch run
for i in range(0,8):
metageneAnalysis("./3.density-data/" + sample[i] + ".swfill.denisty.txt",
"./4.metagene-data/" + sample[i] + ".metagene.txt",
"gene-info.txt")
绘图(R语言):
数据预处理:
library(ggplot2)
library(tidyverse)
library(ggsci)
library(Rmisc)
library(data.table)
###################################################################### start codon
name <- list.files(path = '4.metagene-data/',pattern = '.txt')
map_df(1:length(name), function(x){
tmp = read.table(paste('4.metagene-data/',name[x],sep = ''))
colnames(tmp) <- c('pos','density')
tmp$sample <- sapply(strsplit(name[x],split = '\\.'),'[',1)
tmp$type <- sapply(strsplit(name[x],split = '\\-'),'[',1)
tmp$exp <- sapply(strsplit(name[x],split = '\\-'),'[',2)
return(tmp)
}) %>% data.table() -> df_st
########################## codon position transform
sq <- seq(-51,1500,3);rg <- c(-17:500)
map_df(unique(df_st$sample),function(x){
tmp = df_st[sample %in% x] %>% arrange(pos)
map_df(1:length(sq),function(z){
tmp1 = tmp[pos >= sq[z] & pos <= sq[z] + 2
][,.(mean_norm_density = mean(density)),by = .(type,exp,sample)
][,`:=`(codon_pos = rg[z])]
return(tmp1)
}) -> res
}) -> codon_meta_st
# calculate replicates 95% CI
mean_sd_ci <- codon_meta_st %>%
dplyr::group_by(type,exp,codon_pos) %>%
dplyr::summarise(mean_density = mean(mean_norm_density),
sd = sd(mean_norm_density),
upper = CI(mean_norm_density,ci = 0.95)[1],
lower = CI(mean_norm_density,ci = 0.95)[3])
绘图:
# mean sd
ggplot(mean_sd_ci,aes(x = codon_pos,y = mean_density)) +
geom_ribbon(aes(ymin = mean_density - sd,
ymax = mean_density + sd,
fill = exp),
alpha = 0.5) +
geom_line(aes(color = exp),size = 1) +
geom_hline(yintercept = 1,lty = 'dashed',size = 1,color = 'grey50') +
theme_classic(base_size = 16) +
scale_color_d3(name = '') +
scale_fill_d3(name = '') +
theme(legend.position = 'top',
strip.background = element_rect(color = NA,fill = 'grey')) +
facet_wrap(~type) +
ylab('Mean read density [AU]') +
xlab('Codons / amino acids')
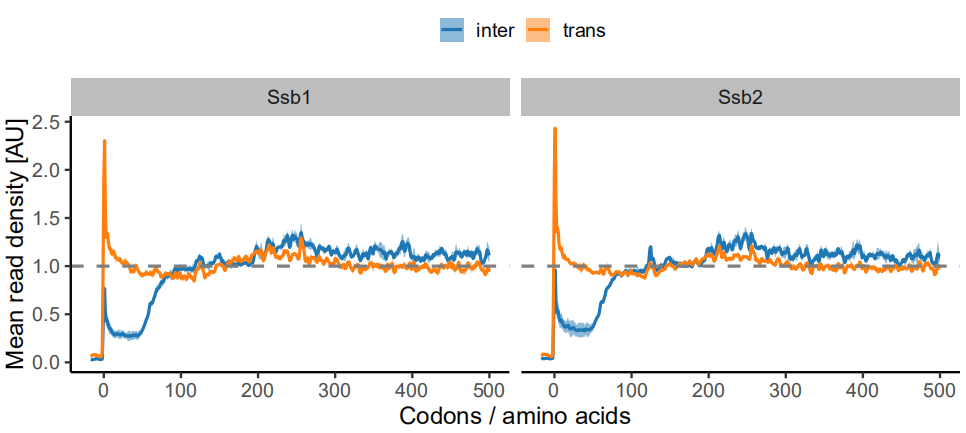
原文
第一次复现结果
6enrichment analysis
这一步可以直接拿上一步已经平滑化好的数据对所有基因计算 ratio (julia代码编写):
function enrichmentAllgenes(geneinfo,ipFile,inputFile,outputFile)
################################################################
# 1. load gene info data
################################################################
# open geneList
geneinfo = open(geneinfo,"r")
# save in dict
geneListDict = Dict{String,Array}()
for line in eachline(geneinfo)
gene_name,startpos,endpos,strand,chr = split(line)
geneListDict[gene_name] = [parse(Int64,startpos),parse(Int64,endpos),strand,chr]
# println(gene_name,startpos,endpos,strand,chr)
# break
end
close(geneinfo)
################################################################
# 2. load interactome and translatome slided window density data(RPM)
################################################################
...
mkdir("5.enrichment-data")
# sample name
IPsample = ["Ssb1-inter-rep1","Ssb2-inter-rep1","Ssb1-inter-rep2","Ssb2-inter-rep2"]
INPUTsample = ["Ssb1-trans-rep1","Ssb2-trans-rep1","Ssb1-trans-rep2","Ssb2-trans-rep2"]
sample = ["Ssb1-rep1","Ssb1-rep2","Ssb2-rep1","Ssb2-rep2"]
# batch run
for i in range(1,4)
enrichmentAllgenes("gene-info.txt",
"./3.density-data/"*IPsample[i]*".swfill.denisty.txt",
"./3.density-data/"*INPUTsample[i]*".swfill.denisty.txt",
"5.enrichment-data/"*sample[i]*".enrichment.txt")
end
绘图(R语言):
数据预处理:
library(ggplot2)
library(tidyverse)
library(ggsci)
library(Rmisc)
library(data.table)
name <- list.files(path = '5.enrichment-data/',pattern = '.txt')
map_df(1:length(name), function(x){
tmp <- fread(paste('5.enrichment-data/',name[x],sep = ''),sep = '\t')
colnames(tmp) <- c('gene_name','pos','ratio')
tmp <- tmp %>% filter(gene_name %in% c('PMT1','CDC37','CCT3','SSC1','PDI1'))
# loop for every gene to process data
map_df(unique(tmp$gene_name),function(y){
tmp1 <- tmp[gene_name == y]
start <- 1
end <- nrow(tmp1)
# 1.transform to codon pos
sq <- seq(1,(end - start + 1),3);rg <- c(1:length(sq))
map_df(1:length(sq),function(z){
tmp2 = tmp1[pos >= sq[z] & pos <= sq[z] + 2
][,.(mean_ratio = mean(ratio)),by = .(gene_name)
][,`:=`(codon_pos = rg[z])]
return(tmp2)
}) -> codon_res
return(codon_res)
}) -> final_res
# add sample info
final_res$sample <- sapply(strsplit(name[x],split = '\\.'),'[',1)
final_res$type <- sapply(strsplit(name[x],split = '\\-'),'[',1)
return(final_res)
}) %>% data.table() -> df_ratio
##################################################
# mean for replicates
merge_rep <- df_ratio %>%
dplyr::group_by(type,gene_name,codon_pos) %>%
dplyr::summarise(mean_rep_ratio = mean(mean_ratio),
mean_sd = sd(mean_ratio))
###################################################
merge_rep$gene_name <- factor(merge_rep$gene_name,
levels = c('PMT1','CDC37','CCT3','SSC1','PDI1'))
绘图:
# plot
ggplot(merge_rep,aes(x = codon_pos,y = mean_rep_ratio)) +
geom_ribbon(aes(ymin = mean_rep_ratio - mean_sd,
ymax = mean_rep_ratio + mean_sd,
fill = type),
alpha = 0.1) +
geom_line(aes(color = type),size = 1) +
geom_hline(yintercept = 1.5,lty = 'dashed',color = 'red',size = 1) +
theme_classic(base_size = 16) +
scale_color_manual(name = '',values = c("Ssb1"="#3F5BDF","Ssb2"="#78B383")) +
scale_fill_manual(name = '',values = c("Ssb1"="#3F5BDF","Ssb2"="#78B383")) +
theme(legend.background = element_blank(),
strip.background = element_rect(color = NA,fill = 'grey')) +
ylab('Mean enrichment [AU] (co-IP/total)') +
xlab('Ribosome position (Codons/amino acids)') +
facet_wrap(~gene_name,scales = 'free',ncol = 3)
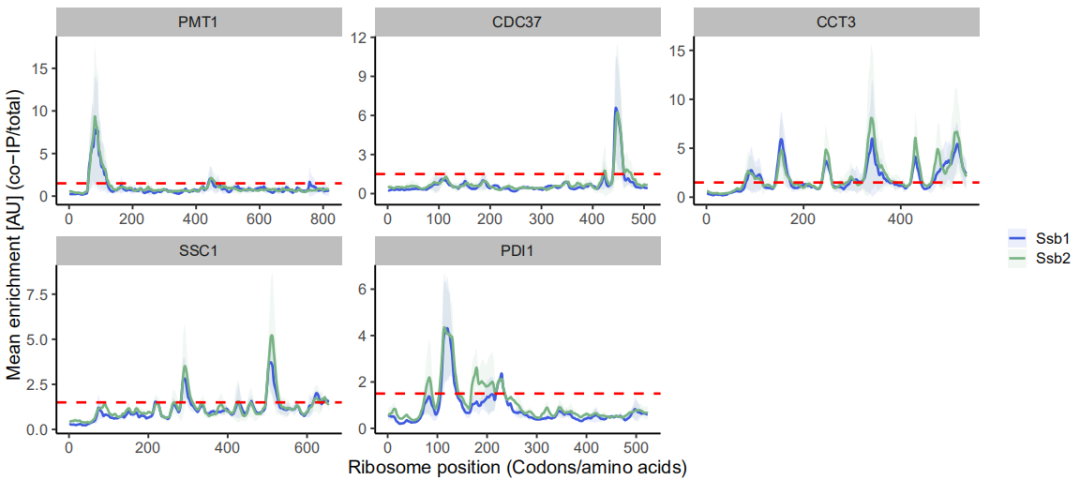
原文
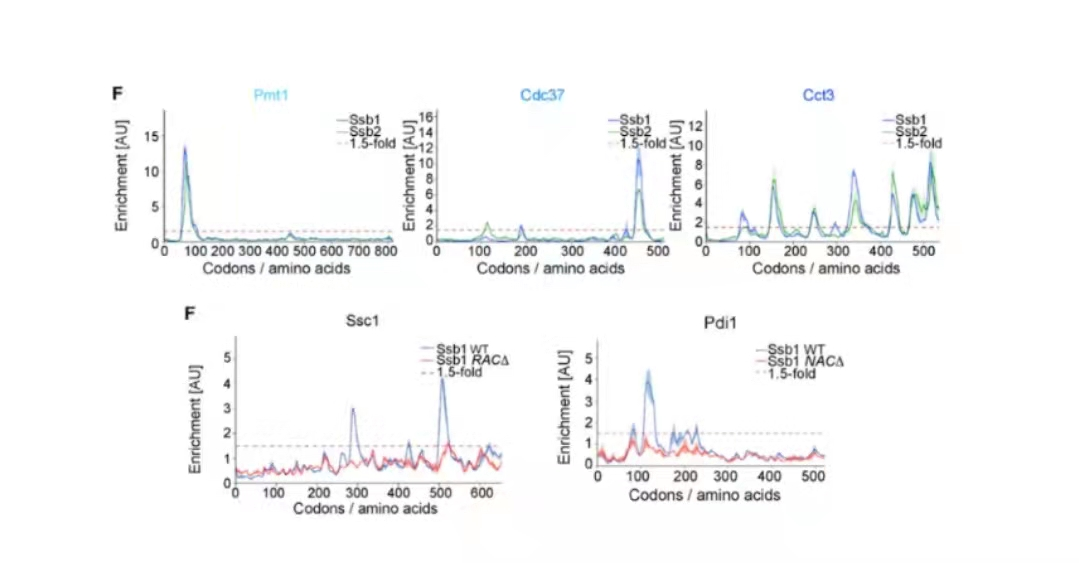
第一次复现结果
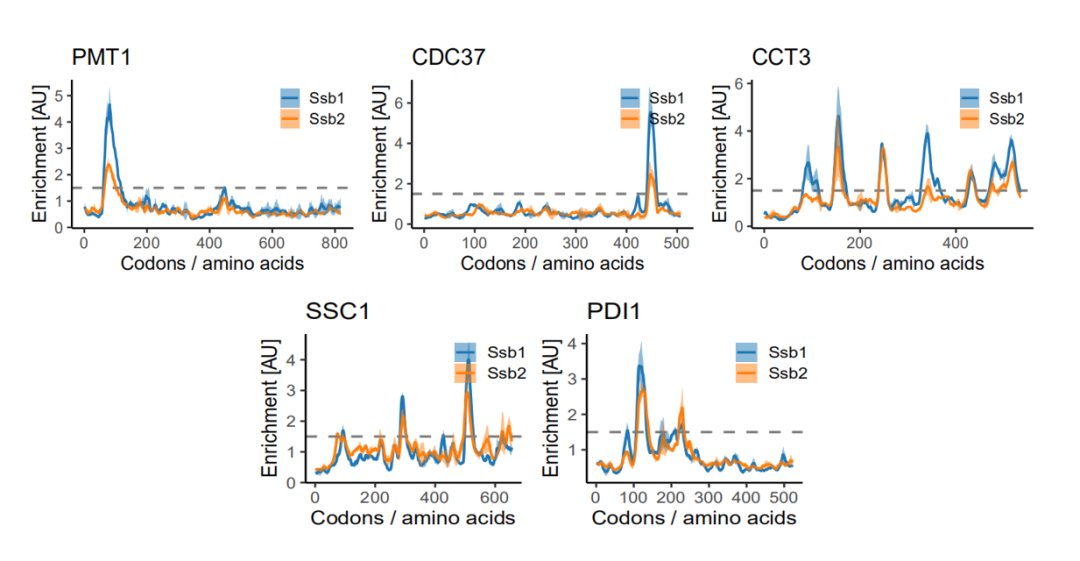
7结尾
也有很多地方需要进一步改进优化,比如减少在 R 里面的数据分析工作。 本文相关的代码适只用于酵母,对于高等真核生物,正如前面提到的挑战,仍需要对代码进行重新编写和测试。 本文探索研究内容纯属个人兴趣,不提供全部代码。

往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集
评论