
谷歌AI一次注释了10%的已知蛋白质序列,超过人类十年研究成果

来源:ScienceAI
编辑 :泽南、张倩
和 AlphaFold 不同,这次谷歌探索的是用深度学习给蛋白质打上功能标签。
蛋白质是组成人体一切细胞、组织的重要成分。机体所有重要的组成部分都需要有蛋白质的参与。
目前已知存在的蛋白质种类有数十亿,但其中大约有三分之一的功能是不可知的。我们迫切地需要探索这片未知区域,因为它们关系到抗菌素耐药性,甚至气候变化等重要议题。例如,青霉素是蛋白质之间自然反应的产物,植物蛋白可用于减少大气中的二氧化碳。
近日,谷歌与欧洲生物信息学研究所合作开发了一种技术 ProtCNN,其能够使用神经网络可靠地预测蛋白质功能,帮助我们缩小蛋白质宇宙中最后不可见的区域。
谷歌表示,这种新方法让我们可以较为准确地预测蛋白质功能、突变的功能效应,并进行蛋白质设计,进而应用于药物发现、酶设计,甚至是了解生命的起源。
论文:《Using deep learning to annotate the protein universe》

论文链接:https://www.nature.com/articles/s41587-021-01179-w
谷歌提出的方法可靠地预测了更多蛋白质的作用,而且它们快速、便宜且易于尝试,其研究已让主流数据库 Pfam 中注释的蛋白质序列增加了近 10%,一举超过了过去十年的增速,并预测了 360 种人类蛋白质功能。
这些结果表明,深度学习模型将成为未来蛋白质注释工具的核心组成部分。
对于大多数人来说,我们更熟悉的是 DeepMind 此前预测蛋白质结构算法 AlphaFold 的工作。AlphaFold 向我们展示了这些神秘生物机器的形状,新研究的重点则是这些机器的作用以及它们的用途。
生物医疗是一个极其活跃的科学领域,每天都有超过十万个蛋白质序列被添加到全球序列数据库中。但是,除非附有功能注释,否则这些条目对从业者的用途非常有限。虽然人们会努力从文献中提取注释,每年评估超过六万篇论文,但这项任务的耗时性质意味着只有 0.03% 的公开可用蛋白质序列是手动注释的。

直接从氨基酸序列推断蛋白质功能是科学社区长久以来一直在研究的方向。从 1980 年代开始,人们就提出了 BLAST 等方法,其依赖于成对的序列比较,假设查询蛋白与已经注释的高度相似的序列具有相同的功能。后来,人们引入了基于 signature 的方法,PROSITE 数据库对在具有特定功能的蛋白质中发现的短氨基酸「基序」进行分类。基于 signature 方法的一个关键改进是开发了 profile 隐马尔可夫模型(pHMM)。这些模型将相关蛋白质序列的对齐折叠成一个模型,该模型为新序列提供似然分数,描述它们与对齐的集合的匹配程度。
在这里至关重要的是,profile HMM 允许更长的 signature 和更模糊的匹配,目前用于更新流行的数据库,如 Interpro 和 Pfam。后期的改进使这些技术更加灵敏,计算效率更高,而它们作为网络工具的高可用性让从业者可以轻松将它们整合到工作流程中去。
这些计算建模方法在学界产生了很大影响。然而,至今仍有三分之一的细菌蛋白质没有被注释出功能。究其原因,当前方法对每个比较序列或模型进行完全独立的比较,因此可能无法充分利用不同功能类共享的特征。
扩展注释的蛋白质序列集需要远程同源检测,即对与训练数据相似度低的序列进行准确分类。新研究得到的基准测试集包含 21,293 个序列。ProtENN 对所有类别分类的准确度显着提高,包括那些具有远距离测试序列的类,这是扩大蛋白质领域覆盖范围的关键要求。为解决从几个例子中推断的挑战,作者使用深度模型学习的序列表示来提高性能。
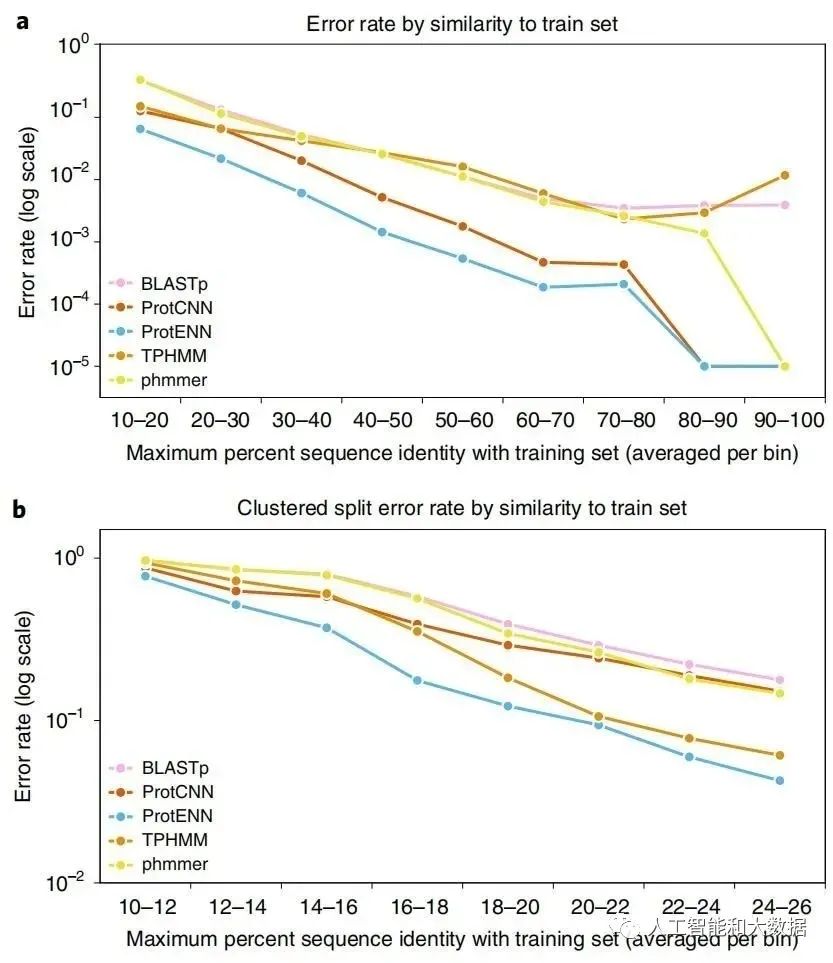
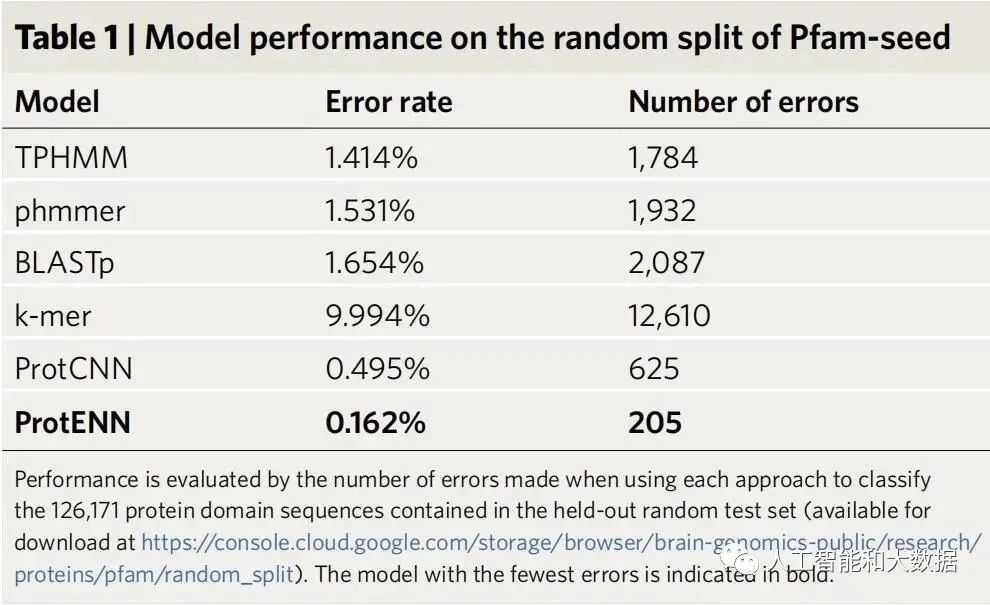
Pfam-seed 模型的性能。
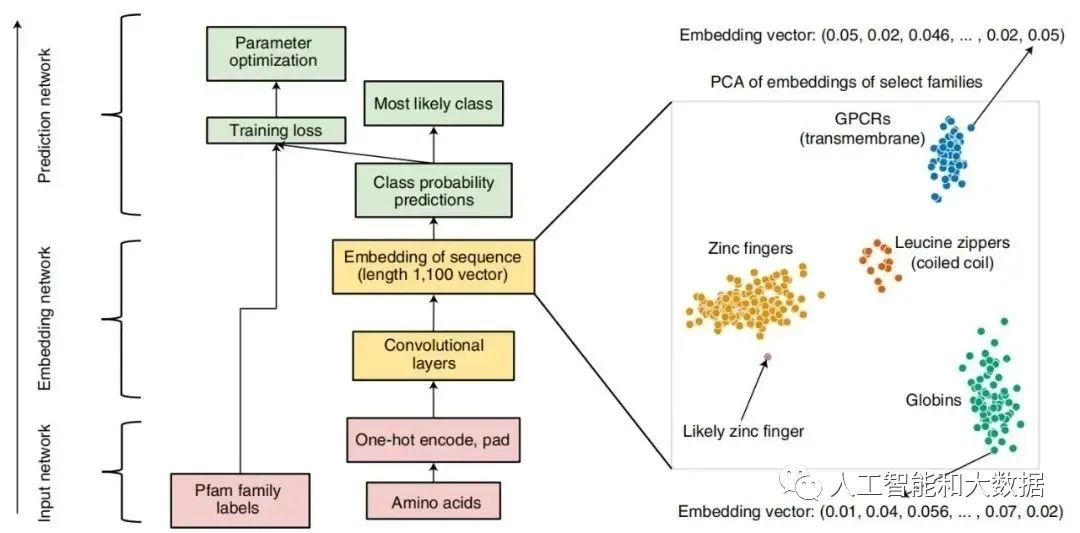
ProtCNN 的架构。中心图展示了输入(红色)、嵌入(黄色)和预测(绿色)网络以及残差网络 ResNet 架构(左),而右图展示了 ProtCNN 和 ProtREP 通过简单的最近邻方法利用。在这一表示中,每个序列对应一个点,来自同一家族的序列通常比来自其他家族的序列更接近。
ProtCNN 学习每序列长度为 1100 的实值向量表示,无论其未对齐长度如何。为获得高精度,来自每个族的表示必须紧密地聚集在一起,以便不同的族很好地相互分离。为了测试这种学习表示是否可用于准确分类最小家族的序列,作者构建了一种称为 ProtREP 的新方法。对于 ProtREP,研究者计算每个家族在其训练序列中的平均学习表示,产生一个标记家族表示。然后通过在学习表示空间中找到其最近的标记来对每个保留的测试序列进行分类。对于相同的计算成本,ProtREP 在聚类分割上超过了 ProtCNN 的准确性。
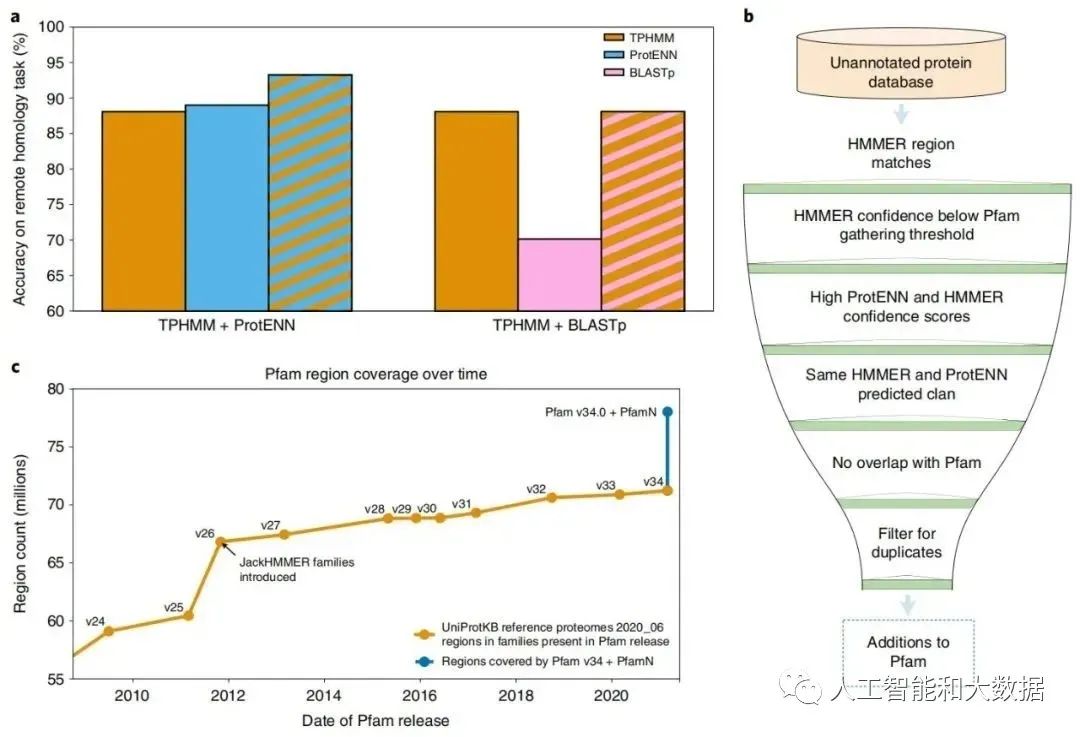
ProtENN 和 TPHMM 的组合提高了远程同源任务的性能。TPHMM 和 ProtENN 模型的简单组合将错误率降低了 38.6%,将 ProtENN 数据的准确度从 89.0% 提高到 93.3%。
为探究深度模型对蛋白质序列数据的了解,作者在来自 Pfam-full 的 80% 的未对齐序列上训练 ProtCNN,并计算了学习氨基酸表示的相似性矩阵。
结果表明,ProtCNN 学习了一种有意义的蛋白质序列表示方式,其可泛化到序列空间未知的部分,可用于预测和理解蛋白质序列的特性。另一个挑战是检测蛋白质结构域及其在蛋白质序列中的位置。此任务类似于图像分割,这正是深度学习模型擅长的任务。虽然 ProtCNN 是使用域进行训练的,但研究展示了 ProtCNN 使用简单的滑动窗口方法将完整序列分割成域的能力。
尽管不使用序列比对,但 ProtCNN 仍显示出了卓越的准确性。