
【行业资讯】谷歌AI一次注释10%蛋白质序列,超人类十年研究成果
预计阅读时间:6分钟

和 AlphaFold 不同,这次谷歌探索的是用深度学习给蛋白质打上功能标签。


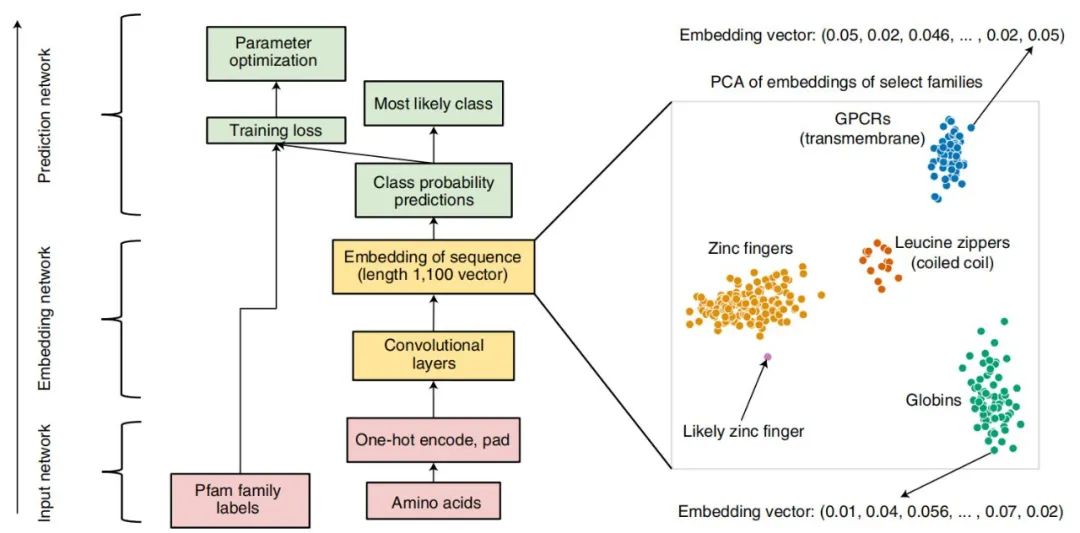
ProtCNN 的架构。中心图展示了输入(红色)、嵌入(黄色)和预测(绿色)网络以及残差网络 ResNet 架构(左),而右图展示了 ProtCNN 和 ProtREP 通过简单的最近邻方法利用。在这一表示中,每个序列对应一个点,来自同一家族的序列通常比来自其他家族的序列更接近
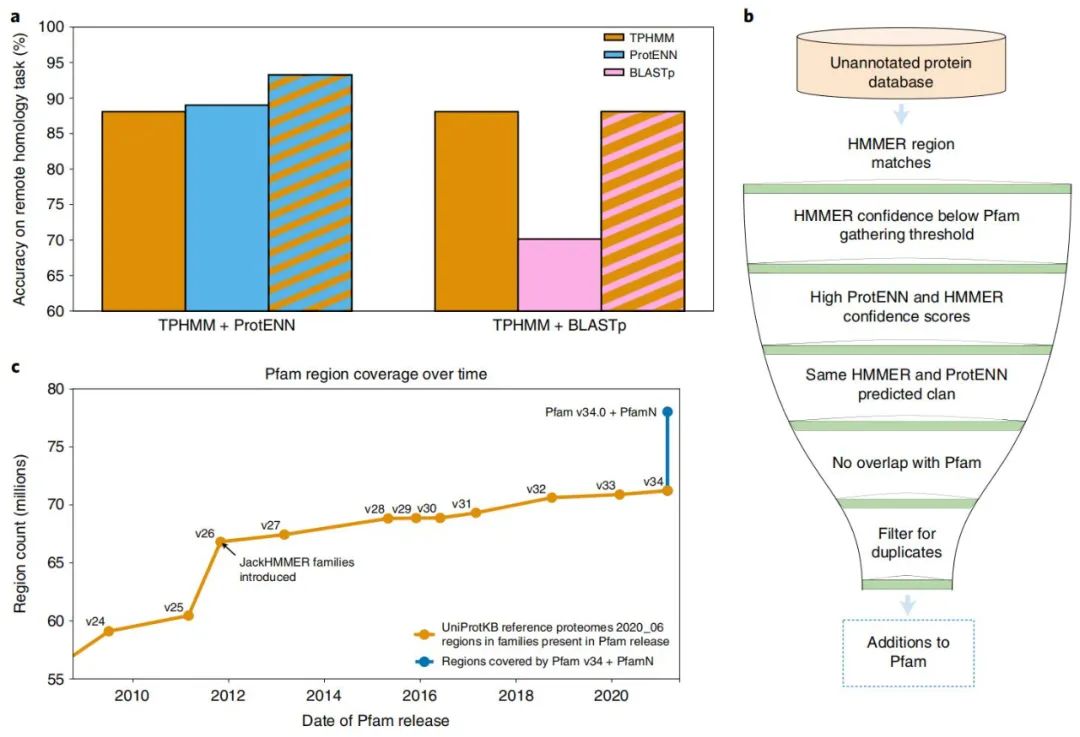
ProtENN 和 TPHMM 的组合提高了远程同源任务的性能。TPHMM 和 ProtENN 模型的简单组合将错误率降低了 38.6%,将 ProtENN 数据的准确度从 89.0% 提高到 93.3%
THE END
来源 | 机器之心
版权声明:本号内容部分来自互联网,转载请注明原文链接和作者,如有侵权或出处有误请和我们联系。
评论