
一文读懂PCA分析 (原理、算法、解释和可视化)
生物信息学习的正确姿势
NGS系列文章包括NGS基础、高颜值在线绘图和分析、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程)、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step))、批次效应处理等内容。

library(knitr)
library(psych)
library(reshape2)
library(ggplot2)
library(ggbeeswarm)
library(scatterplot3d)
library(useful)
library(ggfortify)
mat_show <- function(matr) {
printmrow <- function(x) {
ret <- paste(paste(x,collapse = " & "),"\\\\\n")
sprintf(ret)
}
align_str <- paste0('{',paste0(rep('r',ncol(matr)), collapse=""),'}')
format_mat <- apply(matr,1,printmrow)
add_env <- paste("\\left[\\begin{array}", align_str,
paste(format_mat, collapse=' '),"\\end{array}\\right]")
return(add_env)
}主成分分析简介
主成分分析 (PCA, principal component analysis)是一种数学降维方法,
利用正交变换 (orthogonal
transformation)把一系列可能线性相关的变量转换为一组线性不相关的新变量,也称为主成分,从而利用新变量在更小的维度下展示数据的特征。
主成分是原有变量的线性组合,其数目不多于原始变量。组合之后,相当于我们获得了一批新的观测数据,这些数据的含义不同于原有数据,但包含了之前数据的大部分特征,并且有着较低的维度,便于进一步的分析。
在空间上,PCA可以理解为把原始数据投射到一个新的坐标系统,第一主成分为第一坐标轴,它的含义代表了原始数据中多个变量经过某种变换得到的新变量的变化区间;第二成分为第二坐标轴,代表了原始数据中多个变量经过某种变换得到的第二个新变量的变化区间。这样我们把利用原始数据解释样品的差异转变为利用新变量解释样品的差异。
这种投射方式会有很多,为了最大限度保留对原始数据的解释,一般会用最大方差理论或最小损失理论,使得第一主成分有着最大的方差或变异数
(就是说其能尽量多的解释原始数据的差异);随后的每一个主成分都与前面的主成分正交,且有着仅次于前一主成分的最大方差
(正交简单的理解就是两个主成分空间夹角为90°,两者之间无线性关联,从而完成去冗余操作)。
主成分分析的意义
简化运算。
在问题研究中,为了全面系统地分析问题,我们通常会收集众多的影响因素也就是众多的变量。这样会使得研究更丰富,通常也会带来较多的冗余数据和复杂的计算量。
比如我们我们测序了100种样品的基因表达谱借以通过分子表达水平的差异对这100种样品进行分类。在这个问题中,研究的变量就是不同的基因。每个基因的表达都可以在一定程度上反应样品之间的差异,但某些基因之间却有着调控、协同或拮抗的关系,表现为它们的表达值存在一些相关性,这就造成了统计数据所反映的信息存在一定程度的冗余。另外假如某些基因如持家基因在所有样本中表达都一样,它们对于解释样本的差异也没有意义。这么多的变量在后续统计分析中会增大运算量和计算复杂度,应用PCA就可以在尽量多的保持变量所包含的信息又能维持尽量少的变量数目,帮助简化运算和结果解释。去除数据噪音。
比如说我们在样品的制备过程中,由于不完全一致的操作,导致样品的状态有细微的改变,从而造成一些持家基因也发生了相应的变化,但变化幅度远小于核心基因 (一般认为噪音的方差小于信息的方差)。而PCA在降维的过程中滤去了这些变化幅度较小的噪音变化,增大了数据的信噪比。利用散点图实现多维数据可视化。
在上面的表达谱分析中,假如我们有1个基因,可以在线性层面对样本进行分类;如果我们有2个基因,可以在一个平面对样本进行分类;如果我们有3个基因,可以在一个立体空间对样本进行分类;如果有更多的基因,比如说n个,那么每个样品就是n维空间的一个点,则很难在图形上展示样品的分类关系。利用PCA分析,我们可以选取贡献最大的2个或3个主成分作为数据代表用以可视化。这比直接选取三个表达变化最大的基因更能反映样品之间的差异。(利用Pearson相关系数对样品进行聚类在样品数目比较少时是一个解决办法)发现隐性相关变量。
我们在合并冗余原始变量得到主成分过程中,会发现某些原始变量对同一主成分有着相似的贡献,也就是说这些变量之间存在着某种相关性,为相关变量。同时也可以获得这些变量对主成分的贡献程度。对基因表达数据可以理解为发现了存在协同或拮抗关系的基因。
示例展示原始变量对样品的分类
假设有一套数据集,包含100个样品中某一基因的表达量。如下所示,每一行为一个样品,每一列为基因的表达值。这也是做PCA分析的基本数据组织方式,每一行代表一个样品,每一列代表一组观察数据即一个变量。
count <- 50
Gene1_a <- rnorm(count,5,0.5)
Gene1_b <- rnorm(count,20,0.5)
grp_a <- rep('a', count)
grp_b <- rep('b', count)
cy_data <- data.frame(Gene1 = c(Gene1_a, Gene1_b), Group=c(grp_a, grp_b))
cy_data <- as.data.frame(cy_data)
label <- c(paste0(grp_a, 1:count), paste0(grp_b, 1:count))
row.names(cy_data) <- label
library(knitr)
library(psych)
# Add additional column to data only for plotting
cy_data$Y <- rep(0,count*2)
kable(headTail(cy_data), booktabs=TRUE, caption="Expression profile for Gene1 in 100 samples")| Gene1 | Group | Y | |
|---|---|---|---|
| a1 | 5.99 | a | 0 |
| a2 | 4.66 | a | 0 |
| a3 | 4.92 | a | 0 |
| a4 | 4.79 | a | 0 |
| … | … | NA | … |
| b47 | 20.78 | b | 0 |
| b48 | 20.5 | b | 0 |
| b49 | 20.46 | b | 0 |
| b50 | 19.94 | b | 0 |
从下图可以看出,100个样品根据Gene1表达量的不同在横轴上被被分为了2类,可以看做是在线性水平的分类。
library("ggplot2")
library("ggbeeswarm")
# geom_quasirandom:用于画Jitter Plot
# theme(axis.*.y): 去除Y轴
# xlim, ylim设定坐标轴的区间
ggplot(cy_data,aes(Gene1, Y))+geom_quasirandom(aes(color=factor(Group)), groupOnX=FALSE)+
theme(legend.position=c(0.5,0.7)) + theme(legend.title=element_blank()) +
scale_fill_discrete(name="Group") + theme(axis.line.y=element_blank(),
axis.text.y=element_blank(), axis.ticks.y=element_blank(),
axis.title.y=element_blank()) + ylim(-0.5,5) + xlim(0,25)
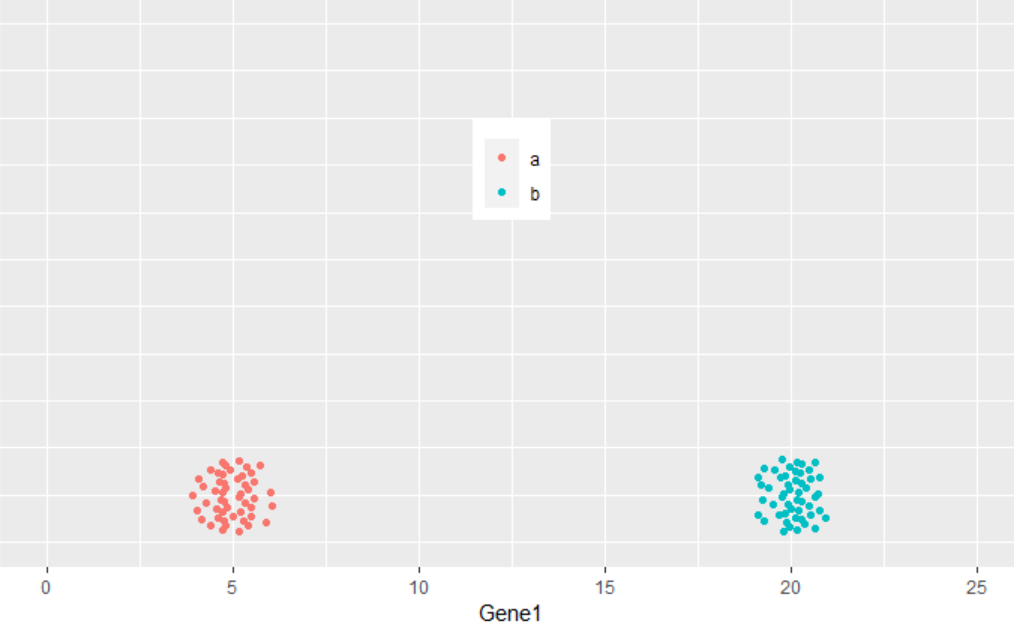
那么如果有2个基因呢?
count <- 50
Gene2_a <- rnorm(count,5,0.2)
Gene2_b <- rnorm(count,5,0.2)
cy_data2 <- data.frame(Gene1 = c(Gene1_a, Gene1_b), Gene2 = c(Gene2_a, Gene2_b),
Group=c(grp_a, grp_b))
cy_data2 <- as.data.frame(cy_data2)
row.names(cy_data2) <- label
kable(headTail(cy_data2), booktabs=T,
caption="Expression profile for Gene1 and Gene2 in 100 samples")| Gene1 | Gene2 | Group | |
|---|---|---|---|
| a1 | 5.99 | 5.35 | a |
| a2 | 4.66 | 5.31 | a |
| a3 | 4.92 | 5.12 | a |
| a4 | 4.79 | 5.11 | a |
| … | … | … | NA |
| b47 | 20.78 | 4.95 | b |
| b48 | 20.5 | 4.9 | b |
| b49 | 20.46 | 4.85 | b |
| b50 | 19.94 | 4.87 | b |
从下图可以看出,100个样品根据Gene1和Gene2的表达量的不同在坐标轴上被被分为了2类,可以看做是在平面水平的分类。而且在这个例子中,我们可以很容易的看出Gene1对样品分类的贡献要比Gene2大,因为Gene1在样品间的表达差异大。
ggplot(cy_data2,aes(Gene1, Gene2))+geom_point(aes(color=factor(Group)))+
theme(legend.position=c(0.5,0.9)) + theme(legend.title=element_blank()) +
ylim(0,10) + xlim(0,25)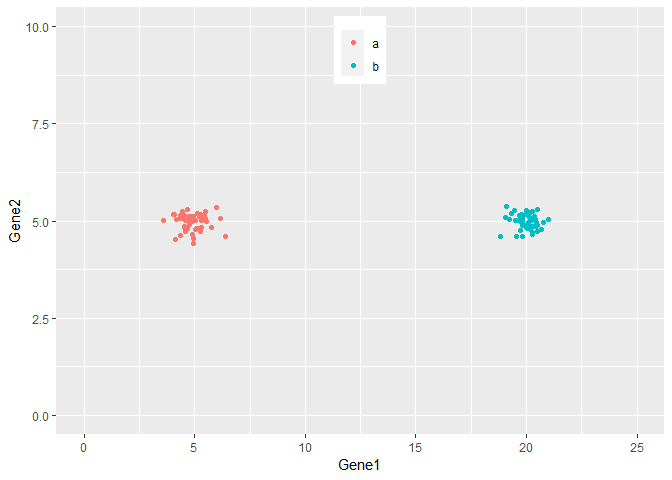
如果有3个基因呢?
count <- 50
Gene3_a <- c(rnorm(count/2,5,0.2), rnorm(count/2,15,0.2))
Gene3_b <- c(rnorm(count/2,15,0.2), rnorm(count/2,5,0.2))
data3 <- data.frame(Gene1 = c(Gene1_a, Gene1_b), Gene2 = c(Gene2_a, Gene2_b),
Gene3 = c(Gene3_a, Gene3_b), Group=c(grp_a, grp_b))
data3 <- as.data.frame(data3)
data3$Group <- as.factor(data3$Group)
row.names(data3) <- label
kable(headTail(data3), booktabs=T, caption="Expression profile for 3 genes in 100 samples")| Gene1 | Gene2 | Gene3 | Group | |
|---|---|---|---|---|
| a1 | 5.99 | 5.35 | 5.14 | a |
| a2 | 4.66 | 5.31 | 5.05 | a |
| a3 | 4.92 | 5.12 | 4.88 | a |
| a4 | 4.79 | 5.11 | 4.8 | a |
| … | … | … | … | NA |
| b47 | 20.78 | 4.95 | 4.91 | b |
| b48 | 20.5 | 4.9 | 5.11 | b |
| b49 | 20.46 | 4.85 | 4.95 | b |
| b50 | 19.94 | 4.87 | 5.18 | b |
从下图可以看出,100个样品根据Gene1、Gene2和Gene3的表达量的不同在坐标轴上被被分为了4类,可以看做是立体空间的分类。而且在这个例子中,我们可以很容易的看出Gene1和Gene3对样品分类的贡献要比Gene2大。
library(scatterplot3d)
colorl <- c("#E69F00", "#56B4E9")
# Extract same number of colors as the Group and same Group would have same color.
colors <- colorl[as.numeric(data3$Group)]
scatterplot3d(data3[,1:3], color=colors, xlim=c(0,25), ylim=c(0,25), zlim=c(0,25),
angle=55, pch=16)
legend("top", legend=levels(data3$Group), col=colorl, pch=16, xpd=T, horiz=T)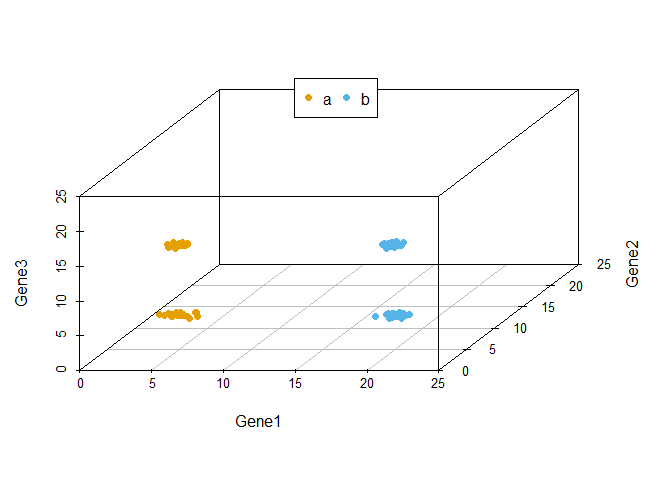
当我们向由Gene1和Gene2构成的X-Y平面做垂线时,可以很明显的看出,Gene2所在的轴对样品的分类没有贡献。因为投射到X-Y屏幕上的点在Y轴几乎处于同一位置。
library(scatterplot3d)
colorl <- c("#E69F00", "#56B4E9")
colors <- colorl[as.numeric(data3$Group)]
scatterplot3d(data3[,1:3], color=colors, xlim=c(0,25), ylim=c(0,25), zlim=c(0,25),type='h')
legend("top", legend=levels(data3$Group), col=colorl, pch=16, xpd=T, horiz=T)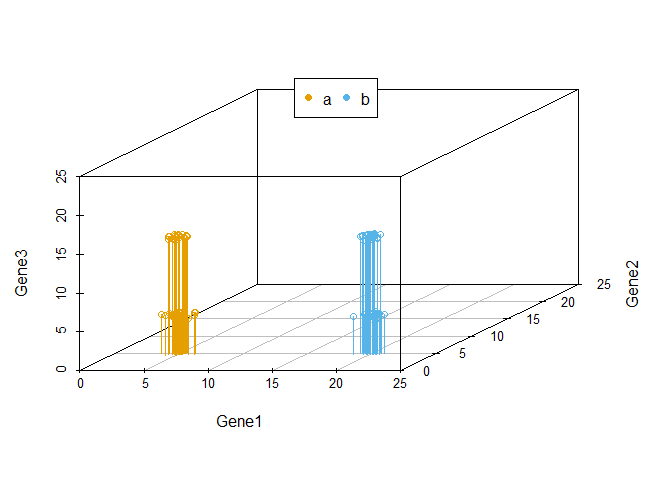
我们把坐标轴做一个转换,可以看到在由Gene1和Gene3构成的X-Y平面上,样品被分为了4类。Gene2对样品的分类几乎没有贡献,因为几乎所有样品在Gene2维度上的值都一样。
library(scatterplot3d)
colorl <- c("#E69F00", "#56B4E9")
colors <- colorl[as.numeric(data3$Group)]
scatterplot3d(x=data3$Gene1, y= data3$Gene3, z= data3$Gene2, color=colors,
xlim=c(0,25), ylim=c(0,25), zlim=c(0,25),type='h')
legend("top", legend=levels(data3$Group), col=colorl, pch=16, xpd=T, horiz=T)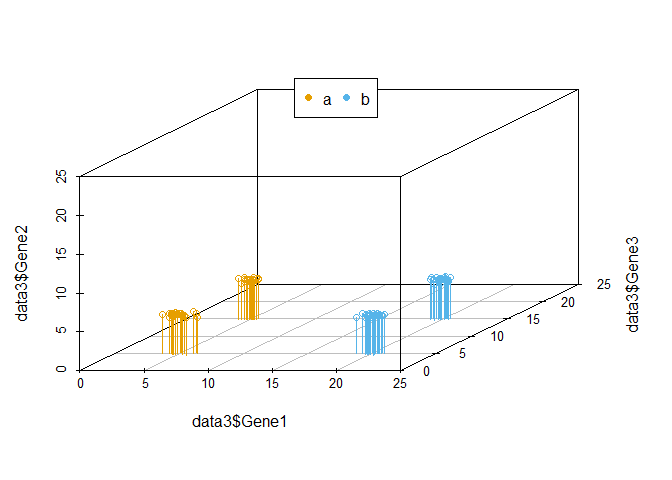
在上述例子中,我们可以很容易的区分出Gene1和Gene3可以作为分类的主成分,而Gene2则对分类没有帮助,可以在计算中去除。
但是如果我们测序了几万个基因的表达时,就很难通过肉眼去看,或者作出一个图供我们筛选哪些基因对样本分类贡献大。这时我们应该怎么做呢?
其中有一个方法是,在这个基因表达矩阵中选出3个变化最大的基因做为3个主成分对样品进行分类。我们试验下效果怎么样。
# 数据集来源于 http://satijalab.org/seurat/old-get-started/
# 原始下载链接 http://www.broadinstitute.org/~rahuls/seurat/seurat_files_nbt.zip
# 为了保证文章的使用,文末附有数据的新下载链接,以防原链接失效
data4 <- read.table("data/HiSeq301-RSEM-linear_values.txt", header=T, row.names=1,sep="\t")
dim(data4)
## [1] 23730 301
library(useful)
kable(corner(data4,r=15,c=8), booktabs=T, caption="Gene expression matrix")| Hi_2338_1 | Hi_2338_2 | Hi_2338_3 | Hi_2338_4 | Hi_2338_5 | Hi_2338_6 | Hi_2338_7 | Hi_2338_8 | |
|---|---|---|---|---|---|---|---|---|
| A1BG | 9.08 | 0.00 | 0.00 | 1.75 | 0.00 | 0.40 | 0.00 | 0.78 |
| A1BG-AS1 | 0.00 | 0.00 | 3.47 | 0.36 | 0.00 | 0.00 | 0.00 | 0.00 |
| A1CF | 0.00 | 0.05 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| A2LD1 | 0.00 | 0.00 | 0.00 | 0.29 | 0.00 | 9.19 | 0.00 | 0.00 |
| A2M | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| A2M-AS1 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| A2ML1 | 0.10 | 0.00 | 0.14 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| A2MP1 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| A4GALT | 0.57 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.35 | 0.00 |
| A4GNT | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| AA06 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| AAAS | 38.95 | 0.00 | 0.00 | 4.44 | 0.00 | 32.90 | 0.00 | 5.58 |
| AACS | 0.12 | 0.00 | 0.00 | 0.00 | 0.58 | 1.03 | 2.16 | 65.74 |
| AACSP1 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| AADAC | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
我们筛选变异系数最大的3个基因。在这之前我们先剔除在少于5个样品中表达的基因和少于1000个表达的基因样品
(这里我们把表达值不小于1的基因视为表达的基因),并把所有基因根据其在不同样品中表达值的变异系数排序。
#去除表达值全为0的行
#data4_nonzero <- data4[rowSums(data4)!=0,]
#筛选符合要求的表达的行和列
#data4_use <- data4[apply(data4,1,function(row) sum(row>=1)>=5),]
#data4_use <- data4[,apply(data4,2,function(col) sum(col>=1)>=1000),]
data4_use <- data4[rowSums(data4>=1)>5,colSums(data4>=1)>1000]
# 对于表达谱数据,因为涉及到PCR的指数扩增,一般会取log处理
# 其它数据log处理会降低数据之间的差异,不一定适用
data4_use_log2 <- log2(data4_use+1)
dim(data4_use_log2)
## [1] 16482 301
# 计算变异系数(标准差除以平均值)度量基因表达变化幅度
#cv <- apply(data4_use_log2,1,sd)/rowMeans(data4_use_log2)
# 根据变异系数排序
#data4_use_log2 <- data4_use_log2[order(cv,decreasing = T),]
# 计算中值绝对偏差 (MAD, median absolute deviation)度量基因表达变化幅度
# 在基因表达中,尽管某些基因很小的变化会导致重要的生物学意义,
# 但是很小的观察值会引入很大的背景噪音,因此也意义不大。
mads <- apply(data4_use_log2, 1, mad)
data4_use_log2 <- data4_use_log2[rev(order(mads)),]
#筛选前3行
data_var3 <- data4_use_log2[1:3,]
# 转置矩阵使得每一行为一个样品,每一列为一组变量
data_var3_forPCA <- t(data_var3)
dim(data_var3_forPCA)
## [1] 301 3
kable(corner(data_var3_forPCA, r=10,c=5), booktabs=TRUE,
caption="A table of the 3 most variable genes")| MT2A | ANXA1 | ARHGDIB | |
|---|---|---|---|
| Hi_2338_1 | 12.211493 | 11.837198 | 9.543283 |
| Hi_2338_2 | 11.306216 | 8.098769 | 8.071623 |
| Hi_2338_3 | 11.926226 | 10.688626 | 9.720535 |
| Hi_2338_4 | 10.974207 | 9.386574 | 9.883376 |
| Hi_2338_5 | 14.603994 | 10.375072 | 9.970379 |
| Hi_2338_6 | 6.904604 | 11.155349 | 10.093510 |
| Hi_2338_7 | 12.436719 | 10.852249 | 7.742882 |
| Hi_2338_8 | 9.798375 | 9.783227 | 6.270716 |
| Hi_2338_9 | 11.743673 | 9.626476 | 9.250251 |
| Hi_2338_10 | 11.240016 | 11.303056 | 0.000000 |
# 获得样品分组信息
sample <- rownames(data_var3_forPCA)
# 把样品名字按 <_> 分割,取出其第二部分作为样品的组名
# lapply(X, FUC) 对列表或向量中每个元素执行FUC操作,FUNC为自定义或R自带的函数
## One better way to generate group
group <- unlist(lapply(strsplit(sample, "_"), function(x) x[2]))
##One way to generate group
#sample_split <- strsplit(sample,"_")
#group <- matrix(unlist(sample_split), ncol=3, byrow=T)[,2]
print(sample[1:4])
## [1] "Hi_2338_1" "Hi_2338_2" "Hi_2338_3" "Hi_2338_4"
print(group[1:4])
## [1] "2338" "2338" "2338" "2338"
data_var3_scatter <- as.data.frame(data_var3_forPCA)
data_var3_scatter$group <- group
kable(corner(data_var3_scatter, r=10,c=5), booktabs=TRUE,
caption="A table of the 3 most variable genes")| MT2A | ANXA1 | ARHGDIB | group | |
|---|---|---|---|---|
| Hi_2338_1 | 12.211493 | 11.837198 | 9.543283 | 2338 |
| Hi_2338_2 | 11.306216 | 8.098769 | 8.071623 | 2338 |
| Hi_2338_3 | 11.926226 | 10.688626 | 9.720535 | 2338 |
| Hi_2338_4 | 10.974207 | 9.386574 | 9.883376 | 2338 |
| Hi_2338_5 | 14.603994 | 10.375072 | 9.970379 | 2338 |
| Hi_2338_6 | 6.904604 | 11.155349 | 10.093510 | 2338 |
| Hi_2338_7 | 12.436719 | 10.852249 | 7.742882 | 2338 |
| Hi_2338_8 | 9.798375 | 9.783227 | 6.270716 | 2338 |
| Hi_2338_9 | 11.743673 | 9.626476 | 9.250251 | 2338 |
| Hi_2338_10 | 11.240016 | 11.303056 | 0.000000 | 2338 |
library(reshape2)
library(ggplot2)
data_var3_melt <- melt(data_var3_scatter, id.vars=c("group"))
kable(corner(data_var3_melt, r=10,c=5), booktabs=TRUE,
caption="A table of the 3 most variable genes in melted format")| group | variable | value |
|---|---|---|
| 2338 | MT2A | 12.211493 |
| 2338 | MT2A | 11.306216 |
| 2338 | MT2A | 11.926226 |
| 2338 | MT2A | 10.974207 |
| 2338 | MT2A | 14.603994 |
| 2338 | MT2A | 6.904604 |
| 2338 | MT2A | 12.436719 |
| 2338 | MT2A | 9.798375 |
| 2338 | MT2A | 11.743673 |
| 2338 | MT2A | 11.240016 |
ggplot(data_var3_melt, aes(factor(variable),value))+ylab("Gene expression")+
geom_violin(aes(fill=factor(group)), stat="ydensity", position="dodge",
scale="width", trim=TRUE) +xlab(NULL)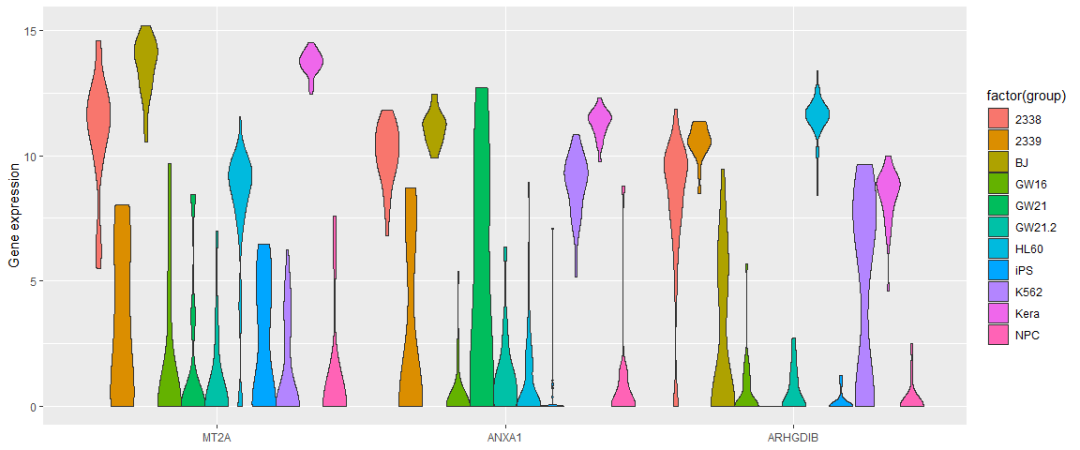
#ggplot(data_var3_melt, aes(factor(variable),value))+ylab("Gene expression")+
#geom_quasirandom(aes(color=factor(group))) +xlab(NULL)
# 根据分组数目确定颜色变量
colorA <- rainbow(length(unique(group)))
# 根据每个样品的分组信息获取对应的颜色变量
colors <- colorA[as.factor(group)]
# 根据样品分组信息获得legend的颜色
colorl <- colorA[as.factor(unique(group))]
# 获得PCH symbol列表
pch_l <- as.numeric(as.factor(unique(group)))
# 产生每个样品的pch symbol
pch <- pch_l[as.factor(group)]
scatterplot3d(data_var3_forPCA[,1:3], color=colors, pch=pch)
legend(0,10, legend=levels(as.factor(group)), col=colorl, pch=pch_l, xpd=T, horiz=F, ncol=6)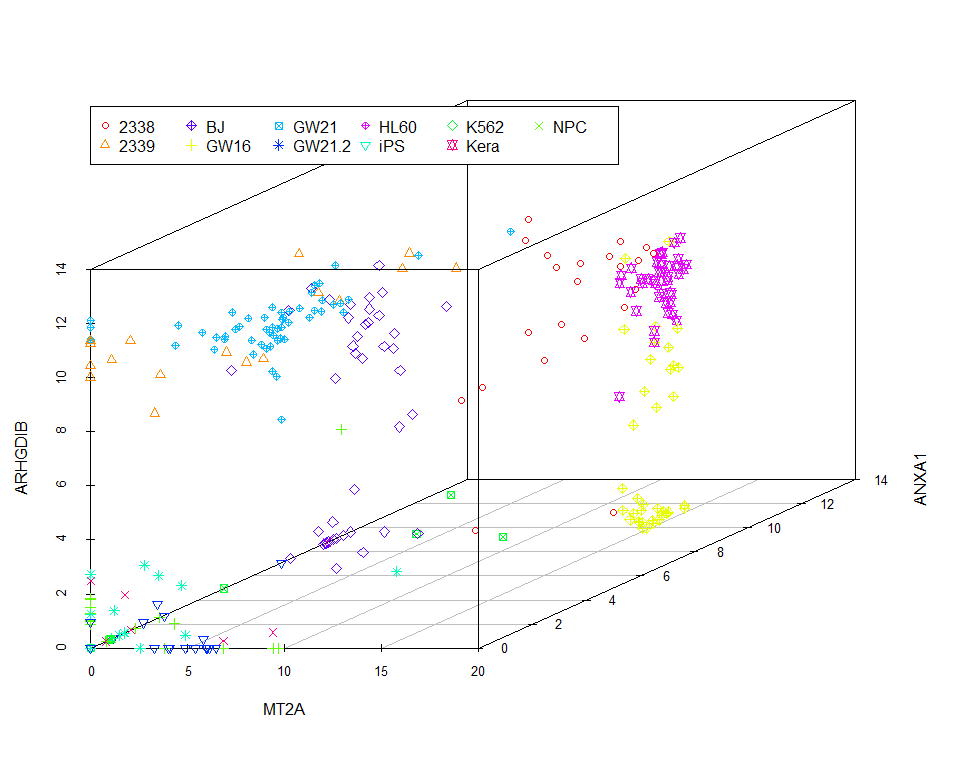
我们看到图中的样品并没有按照预先设定的标签完全分开。当然我们也可以通过其他方法筛选变异最大的三个基因,最终的分类效果不会相差很大。因为不管怎么筛选,我们都只用到了3个基因的表达量。
假如我们把这个数据用PCA来分类,结果是怎样的呢?
# Pay attention to the format of PCA input
# Rows are samples and columns are variables
data4_use_log2_t <- t(data4_use_log2)
# Add group column for plotting
data4_use_log2_label <- as.data.frame(data4_use_log2_t)
data4_use_log2_label$group <- group
# By default, prcomp will centralized the data using mean.
# Normalize data for PCA by dividing each data by column standard deviation.
# Often, we would normalize data.
# Only when we care about the real number changes other than the trends,
# `scale` can be set to TRUE.
# We will show the differences of scaling and un-scaling effects.
pca <- prcomp(data4_use_log2_t, scale=T)
# sdev: standard deviation of the principle components.
# Square to get variance
percentVar <- pca$sdev^2 / sum( pca$sdev^2)
# To check what's in pca
print(str(pca))
## List of 5
## $ sdev : num [1:301] 36.6 30.4 23.3 21.6 19.9 ...
## $ rotation: num [1:16482, 1:301] -0.01133 -0.01955 -0.00199 -0.00569 -0.0204 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:16482] "MT2A" "ANXA1" "ARHGDIB" "RND3" ...
## .. ..$ : chr [1:301] "PC1" "PC2" "PC3" "PC4" ...
## $ center : Named num [1:16482] 6.5 5.47 5.43 4.23 4.1 ...
## ..- attr(*, "names")= chr [1:16482] "MT2A" "ANXA1" "ARHGDIB" "RND3" ...
## $ scale : Named num [1:16482] 5.62 4.96 4.78 4.13 3.98 ...
## ..- attr(*, "names")= chr [1:16482] "MT2A" "ANXA1" "ARHGDIB" "RND3" ...
## $ x : num [1:301, 1:301] -37.6 -28.6 -30.6 -43.7 -11.4 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:301] "Hi_2338_1" "Hi_2338_2" "Hi_2338_3" "Hi_2338_4" ...
## .. ..$ : chr [1:301] "PC1" "PC2" "PC3" "PC4" ...
## - attr(*, "class")= chr "prcomp"
## NULL从图中可以看到,数据呈现了一定的分类模式
(当然这个分类结果也不理想,我们随后再进一步优化)。
library(ggfortify)
autoplot(pca, data=data4_use_log2_label, colour="group") +
xlab(paste0("PC1 (", round(percentVar[1]*100), "% variance)")) +
ylab(paste0("PC2 (", round(percentVar[2]*100), "% variance)")) + theme_bw() +
theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank()) +
theme(legend.position="right")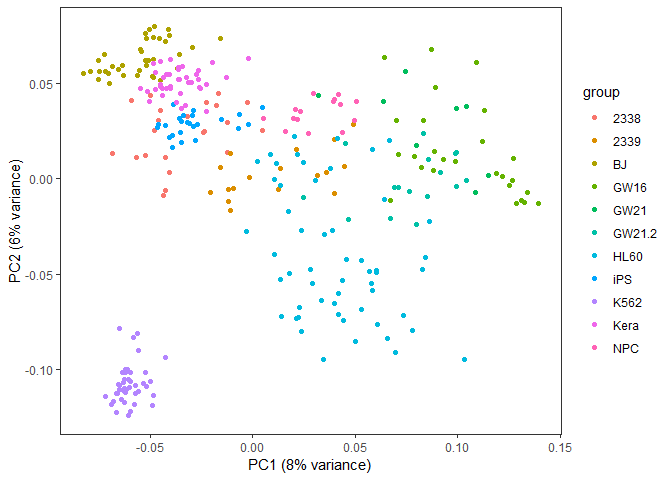
采用3个主成分获得的分类效果优于2个主成分,因为这样保留的原始信息更多。
# 根据分组数目确定颜色变量
colorA <- rainbow(length(unique(group)))
# 根据每个样品的分组信息获取对应的颜色变量
colors <- colorA[as.factor(group)]
# 根据样品分组信息获得legend的颜色
colorl <- colorA[as.factor(unique(group))]
# 获得PCH symbol列表
pch_l <- as.numeric(as.factor(unique(group)))
# 产生每个样品的pch symbol
pch <- pch_l[as.factor(group)]
pc <- as.data.frame(pca$x)
scatterplot3d(x=pc$PC1, y=pc$PC2, z=pc$PC3, pch=pch, color=colors,
xlab=paste0("PC1 (", round(percentVar[1]*100), "% variance)"),
ylab=paste0("PC2 (", round(percentVar[2]*100), "% variance)"),
zlab=paste0("PC3 (", round(percentVar[3]*100), "% variance)"))
legend(-3,8, legend=levels(as.factor(group)), col=colorl, pch=pch_l, xpd=T, horiz=F, ncol=6)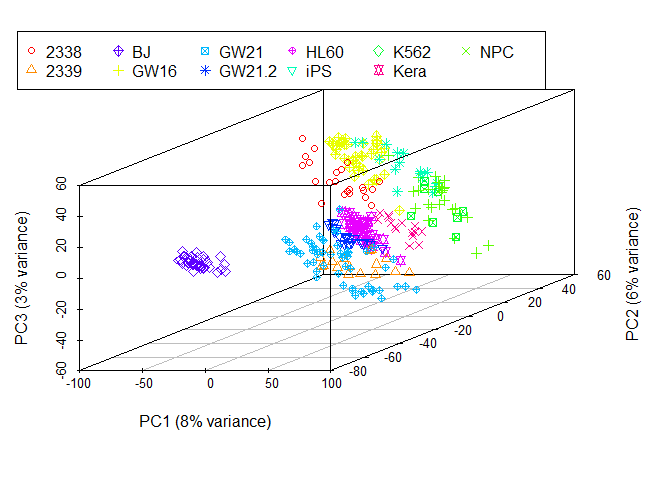
PCA的实现原理
在上面的例子中,PCA分析不是简单地选取2个或3个变化最大的基因,而是先把原始的变量做一个评估,计算各个变量各自的变异度(方差)和两两变量的相关度(协方差),得到一个协方差矩阵。在这个协方差矩阵中,对角线的值为每一个变量的方差,其它值为每两个变量的协方差。随后对原变量的协方差矩阵对角化处理,即求解其特征值和特征向量。原变量与特征向量的乘积(对原始变量的线性组合)即为新变量(回顾下线性代数中的矩阵乘法);新变量的协方差矩阵为对角协方差矩阵且对角线上的方差由大到小排列;然后从新变量中选择信息最丰富也就是方差最大的的前2个或前3个新变量也就是主成分用以可视化。下面我们一步步阐释这是怎么做的。
我们先回忆两个数学概念,方差和协方差。方差用来表示一组一维数据的离散程度。协方差表示2组一维数据的相关性。当协方差为0时,表示两组数据完全独立。当协方差为正时,表示一组数据增加时另外一组也会增加;当协方差为负时表示一组数据增加时另外一组数据会降低
(与相关系数类似)。如果我们有很多组一维数据,比如很多基因的表达数据,就会得到很多协方差,这就构成了协方差矩阵。

假如我们有一个矩阵如下,
mat <- as.data.frame(matrix(rnorm(20,0,1), nrow=4))
colnames(mat) <- paste0("Gene_", letters[1:5])
rownames(mat) <- paste0("Samp_", 1:4)
mat
## Gene_a Gene_b Gene_c Gene_d Gene_e
## Samp_1 -1.2207554 0.7608654 -0.2921542 0.2781680 -0.5515104
## Samp_2 -0.3855339 -1.3785382 1.1008845 0.5385477 0.1083430
## Samp_3 0.5828754 0.7443152 0.2221331 1.0248715 0.2456042
## Samp_4 -1.6524972 -1.6259796 0.8235352 -0.7186743 0.3295052平均值中心化 (mean centering):中心化数据使其平均值为0
# mean-centering data for columns
# Get mean-value matrix first
mat_mean_norm <- mat - rep(colMeans(mat),rep.int(nrow(mat),ncol(mat)))
mat_mean_norm
## Gene_a Gene_b Gene_c Gene_d Gene_e
## Samp_1 -0.5517776 1.135700 -0.7557539 -0.002560247 -0.58449593
## Samp_2 0.2834438 -1.003704 0.6372848 0.257819506 0.07535752
## Samp_3 1.2518532 1.119149 -0.2414665 0.744143242 0.21261872
## Samp_4 -0.9835194 -1.251145 0.3599356 -0.999402500 0.29651969
# mean-centering using scale for columns
scale(mat, center=T, scale=F)
## Gene_a Gene_b Gene_c Gene_d Gene_e
## Samp_1 -0.5517776 1.135700 -0.7557539 -0.002560247 -0.58449593
## Samp_2 0.2834438 -1.003704 0.6372848 0.257819506 0.07535752
## Samp_3 1.2518532 1.119149 -0.2414665 0.744143242 0.21261872
## Samp_4 -0.9835194 -1.251145 0.3599356 -0.999402500 0.29651969
## attr(,"scaled:center")
## Gene_a Gene_b Gene_c Gene_d Gene_e
## -0.66897778 -0.37483430 0.46359965 0.28072824 0.03298553中位数中心化 (median
centering):如果数据变换范围很大或有异常值,中位数标准化效果会更好。
# median-centering data for columns
mat_median_norm <- mat - rep(apply(mat,2,median),rep.int(nrow(mat),ncol(mat)))
mat_mean_norm
## Gene_a Gene_b Gene_c Gene_d Gene_e
## Samp_1 -0.5517776 1.135700 -0.7557539 -0.002560247 -0.58449593
## Samp_2 0.2834438 -1.003704 0.6372848 0.257819506 0.07535752
## Samp_3 1.2518532 1.119149 -0.2414665 0.744143242 0.21261872
## Samp_4 -0.9835194 -1.251145 0.3599356 -0.999402500 0.29651969我们可以计算Gene_a的方差为 0.973082
(var(mat$Gene_a));Gene_a和Gene_b的协方差为0.5734631。
mat中5组基因的表达值的方差计算如下:
apply(mat,2,var)
## Gene_a Gene_b Gene_c Gene_d Gene_e
## 0.9730820 1.7050318 0.3883852 0.5396773 0.1601483mat中5组基因表达值的协方差计算如下:
cov(mat)
## Gene_a Gene_b Gene_c Gene_d Gene_e
## Gene_a 0.97308195 0.5734631 -0.01954727 0.66299331 0.10613531
## Gene_b 0.57346308 1.7050318 -0.73950788 0.60717437 -0.29082853
## Gene_c -0.01954727 -0.7395079 0.38838520 -0.12438895 0.18171565
## Gene_d 0.66299331 0.6071744 -0.12438895 0.53967732 -0.03906622
## Gene_e 0.10613531 -0.2908285 0.18171565 -0.03906622 0.16014830如果均值为0,数值矩阵的协方差矩阵为矩阵的乘积
(实际上是矩阵的转置与其本身的乘积除以变量的维数减1)。
# Covariance matrix for Mean normalized matrix
cov(mat_mean_norm)
## Gene_a Gene_b Gene_c Gene_d Gene_e
## Gene_a 0.97308195 0.5734631 -0.01954727 0.66299331 0.10613531
## Gene_b 0.57346308 1.7050318 -0.73950788 0.60717437 -0.29082853
## Gene_c -0.01954727 -0.7395079 0.38838520 -0.12438895 0.18171565
## Gene_d 0.66299331 0.6071744 -0.12438895 0.53967732 -0.03906622
## Gene_e 0.10613531 -0.2908285 0.18171565 -0.03906622 0.16014830
# Covariance matrix for Mean normalized matrix
# crossprod: matrix multiplication
crossprod(as.matrix(mat_mean_norm)) / (nrow(mat_mean_norm)-1)
## Gene_a Gene_b Gene_c Gene_d Gene_e
## Gene_a 0.97308195 0.5734631 -0.01954727 0.66299331 0.10613531
## Gene_b 0.57346308 1.7050318 -0.73950788 0.60717437 -0.29082853
## Gene_c -0.01954727 -0.7395079 0.38838520 -0.12438895 0.18171565
## Gene_d 0.66299331 0.6071744 -0.12438895 0.53967732 -0.03906622
## Gene_e 0.10613531 -0.2908285 0.18171565 -0.03906622 0.16014830
# Use %*% for matrix multiplication (slower)
t(as.matrix(mat_mean_norm)) %*% as.matrix(mat_mean_norm) / (nrow(mat_mean_norm)-1)
## Gene_a Gene_b Gene_c Gene_d Gene_e
## Gene_a 0.97308195 0.5734631 -0.01954727 0.66299331 0.10613531
## Gene_b 0.57346308 1.7050318 -0.73950788 0.60717437 -0.29082853
## Gene_c -0.01954727 -0.7395079 0.38838520 -0.12438895 0.18171565
## Gene_d 0.66299331 0.6071744 -0.12438895 0.53967732 -0.03906622
## Gene_e 0.10613531 -0.2908285 0.18171565 -0.03906622 0.16014830用矩阵形式书写如下,便于理解
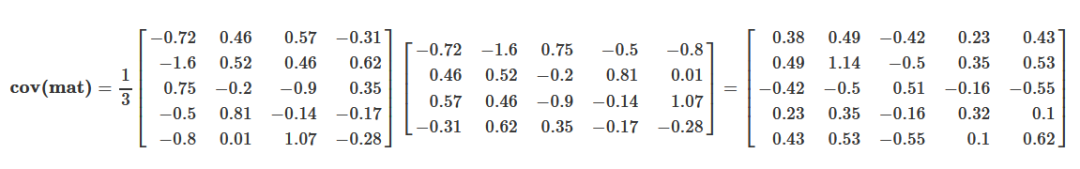
根据前面的描述,原始变量的协方差矩阵表示原始变量自身的方差(协方差矩阵的主对角线位置)和原始变量之间的相关程度(非主对角线位置)。如果从这些数据中筛选主成分,则要选择方差大(主对角线值大),且与其它已选变量之间相关性最小的变量(非主对角线值很小)。如果这些原始变量之间毫不相关,则它们的协方差矩阵在除主对角线处外其它地方的值都为0,这种矩阵成为对角矩阵。
而做PCA分析就是产生一组新的变量,使得新变量的协方差矩阵为对角阵,满足上面的要求。从而达到去冗余的目的。然后再选取方差大的变量,实现降维和去噪。
如果正向推导,这种组合可能会有很多种,一一计算会比较麻烦。那反过来看呢?
我们不去寻找这种组合,而是计算如何使原变量的协方差矩阵变为对角阵。
数学推导中谨记的两个概念:
假设: 把未求解到的变量假设出来,用符号代替;这样有助于思考和演算
逆向:如果正向推导求不出,不妨倒着来;尽量多的利用已有信息
前面提到,新变量(Ym, k)是原始变量(Xm, n)(原始变量的协方差矩阵为(Cn, n))的线性组合,那么假设我们找到了这么一个线性组合(命名为特征矩阵(Pn, k)),得到一组新变量Ym, k = Xm, nPn, k,并且新变量的协方差矩阵(Dk, k)为对角阵。那么这个特征矩阵(Pn, k)需要符合什么条件呢?
从矩阵运算可以看出,最终的特征矩阵(Pn, k)需要把原变量协方差矩阵(Cn, n)转换为对角阵(因为新变量的协方差矩阵(Dk, k)为对角阵),并且对角元素从大到小排列(保证每个主成分的贡献度依次降低)。
现在就把求解新变量的任务转变为了求解原变量协方差矩阵的对角化问题了。在线性代数中,矩阵对角化的问题就是求解矩阵的特征值和特征向量的问题。
我们举一个例子讲述怎么求解特征值和特征向量。
假设An, n为n阶对称阵,如存在λ和非零向量x,使得A**x = λ**x,则称λ为矩阵An, n的特征值,非零向量x为为矩阵An, n对应于特征值λ的特征向量。
根据这个定义可以得出(An, n − λ**E)x = 0,由于x为非零向量,所以行列式|A − λ**E| = 0。
由此求解出n个根λ1, λ2, …, λ3就是矩阵A的特征值。
回顾下行列式的计算:
行列式的值为行列式第一列的每一个数乘以它的余子式(余子式是行列式中除去当前元素所在行和列之后剩下的行列式)。
当行列式中存在线性相关的行或列或者有一行或一列元素全为0时,行列式的值为0。
上三角形行列式的值为其主对角线上元素的乘积。
互换行列式的两行或两列,行列式变号。
行列式的某一列(行)乘以同意书加到另一列(列)对应元素上去,行列式不变。

简单的PCA实现
我们使用前面用到的数据data3来演示下如何用R函数实现PCA的计算,并与R中自带的prcomp做个比较。
library(knitr)
kable(headTail(data3), booktabs=T, caption="Expression profile for 3 genes in 100 samples")| Gene1 | Gene2 | Gene3 | Group | |
|---|---|---|---|---|
| a1 | 5.99 | 5.35 | 5.14 | a |
| a2 | 4.66 | 5.31 | 5.05 | a |
| a3 | 4.92 | 5.12 | 4.88 | a |
| a4 | 4.79 | 5.11 | 4.8 | a |
| … | … | … | … | NA |
| b47 | 20.78 | 4.95 | 4.91 | b |
| b48 | 20.5 | 4.9 | 5.11 | b |
| b49 | 20.46 | 4.85 | 4.95 | b |
| b50 | 19.94 | 4.87 | 5.18 | b |
标准化数据
data3_center_scale <- scale(data3[,1:3], center=T, scale=T)
kable(headTail(data3_center_scale), booktabs=T,
caption="Normalized expression for 3 genes in 100 samples")| Gene1 | Gene2 | Gene3 | |
|---|---|---|---|
| a1 | -0.85 | 1.83 | -0.96 |
| a2 | -1.03 | 1.66 | -0.98 |
| a3 | -0.99 | 0.72 | -1.01 |
| a4 | -1.01 | 0.67 | -1.03 |
| … | … | … | … |
| b47 | 1.09 | -0.11 | -1.01 |
| b48 | 1.06 | -0.38 | -0.97 |
| b49 | 1.05 | -0.61 | -1 |
| b50 | 0.98 | -0.52 | -0.95 |
计算协方差矩阵
data3_center_scale_cov <- cov(data3_center_scale)
kable(data3_center_scale_cov, booktabs=T,
caption="Covariance matrix for 3 genes in 100 samples")| Gene1 | Gene2 | Gene3 | |
|---|---|---|---|
| Gene1 | 1.0000000 | 0.0255834 | -0.0087919 |
| Gene2 | 0.0255834 | 1.0000000 | 0.0553298 |
| Gene3 | -0.0087919 | 0.0553298 | 1.0000000 |
求解特征值和特征向量
data3_center_scale_cov_eigen <- eigen(data3_center_scale_cov)
# 特征值,从大到小排序
data3_center_scale_cov_eigen$values
## [1] 1.0580005 1.0066389 0.9353605
# 特征向量, 每一列为对应特征值的特征向量
data3_center_scale_cov_eigen$vectors
## [,1] [,2] [,3]
## [1,] 0.2192050 0.90784568 0.3574429
## [2,] 0.7223522 0.09525968 -0.6849327
## [3,] 0.6558631 -0.40834033 0.6349030产生新的矩阵
pc_select = 3
label = paste0("PC",c(1:pc_select))
data3_new <- data3_center_scale %*% data3_center_scale_cov_eigen$vectors[,1:pc_select]
colnames(data3_new) <- label
kable(headTail(data3_new), booktabs=T,
caption="PCA generated matrix for the expression of 3 genes in 100 samples")| PC1 | PC2 | PC3 | |
|---|---|---|---|
| a1 | 0.51 | -0.21 | -2.17 |
| a2 | 0.33 | -0.37 | -2.12 |
| a3 | -0.36 | -0.42 | -1.49 |
| a4 | -0.41 | -0.43 | -1.47 |
| … | … | … | … |
| b47 | -0.5 | 1.39 | -0.17 |
| b48 | -0.68 | 1.32 | 0.03 |
| b49 | -0.86 | 1.31 | 0.16 |
| b50 | -0.79 | 1.23 | 0.1 |
比较原始数据和新产生的主成分对样品的聚类
#library(scatterplot3d)
colorl <- c("#E69F00", "#56B4E9")
# Extract same number of colors as the Group and same Group would have same color.
colors <- colorl[as.numeric(data3$Group)]
# 1 row 2 columns
par(mfrow=c(1,2))
scatterplot3d(data3[,1:3], color=colors, angle=55, pch=16, main="Original data")
legend("top", legend=levels(data3$Group), col=colorl, pch=16, xpd=T, horiz=T)
scatterplot3d(data3_new, color=colors,angle=55, pch=16, main="Principle components")
legend("top", legend=levels(data3$Group), col=colorl, pch=16, xpd=T, horiz=T)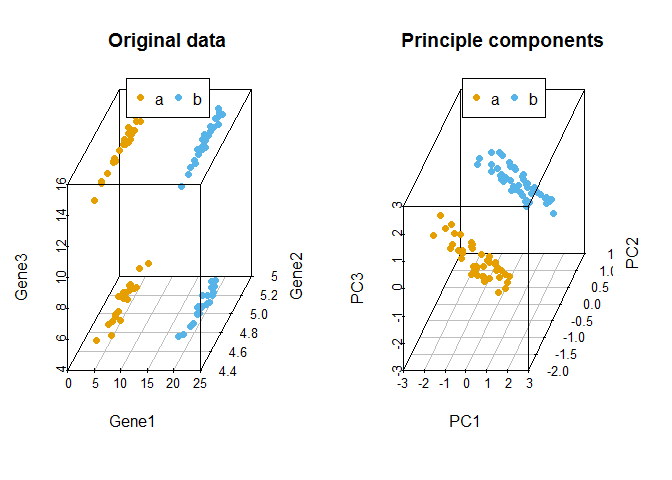
#par(mfrow=c(1,1))利用prcomp进行主成分分析
pca_data3 <- prcomp(data3[,1:3], center=TRUE, scale=TRUE)
#Show whats in the result returned by prcomp
str(pca_data3)
## List of 5
## $ sdev : num [1:3] 1.029 1.003 0.967
## $ rotation: num [1:3, 1:3] 0.2192 0.7224 0.6559 -0.9078 -0.0953 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:3] "Gene1" "Gene2" "Gene3"
## .. ..$ : chr [1:3] "PC1" "PC2" "PC3"
## $ center : Named num [1:3] 12.46 4.98 10
## ..- attr(*, "names")= chr [1:3] "Gene1" "Gene2" "Gene3"
## $ scale : Named num [1:3] 7.602 0.202 5.06
## ..- attr(*, "names")= chr [1:3] "Gene1" "Gene2" "Gene3"
## $ x : num [1:100, 1:3] 0.506 0.331 -0.36 -0.409 -0.27 ...
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:100] "a1" "a2" "a3" "a4" ...
## .. ..$ : chr [1:3] "PC1" "PC2" "PC3"
## - attr(*, "class")= chr "prcomp"
# 新的数据,与前面计算的抑制
data3_pca_new <- pca_data3$x
kable(headTail(data3_pca_new), booktabs=T,
caption="PCA generated matrix usig princomp for the expression of 3 genes in 100 samples")| PC1 | PC2 | PC3 | |
|---|---|---|---|
| a1 | 0.51 | 0.21 | -2.17 |
| a2 | 0.33 | 0.37 | -2.12 |
| a3 | -0.36 | 0.42 | -1.49 |
| a4 | -0.41 | 0.43 | -1.47 |
| … | … | … | … |
| b47 | -0.5 | -1.39 | -0.17 |
| b48 | -0.68 | -1.32 | 0.03 |
| b49 | -0.86 | -1.31 | 0.16 |
| b50 | -0.79 | -1.23 | 0.1 |
# 特征向量,与我们前面计算的一致(特征向量的符号是任意的)
pca_data3$rotation
## PC1 PC2 PC3
## Gene1 0.2192050 -0.90784568 0.3574429
## Gene2 0.7223522 -0.09525968 -0.6849327
## Gene3 0.6558631 0.40834033 0.6349030比较手动实现的PCA与prcomp实现的PCA的聚类结果
#library(scatterplot3d)
colorl <- c("#E69F00", "#56B4E9")
# Extract same number of colors as the Group and same Group would have same color.
colors <- colorl[as.numeric(data3$Group)]
# 1 row 2 columns
par(mfrow=c(1,2))
scatterplot3d(data3_new, color=colors,angle=55, pch=16, main="PCA by steps")
legend("top", legend=levels(data3$Group), col=colorl, pch=16, xpd=T, horiz=T)
scatterplot3d(data3_pca_new, color=colors,angle=55, pch=16, main="PCA by prcomp")
legend("top", legend=levels(data3$Group), col=colorl, pch=16, xpd=T, horiz=T)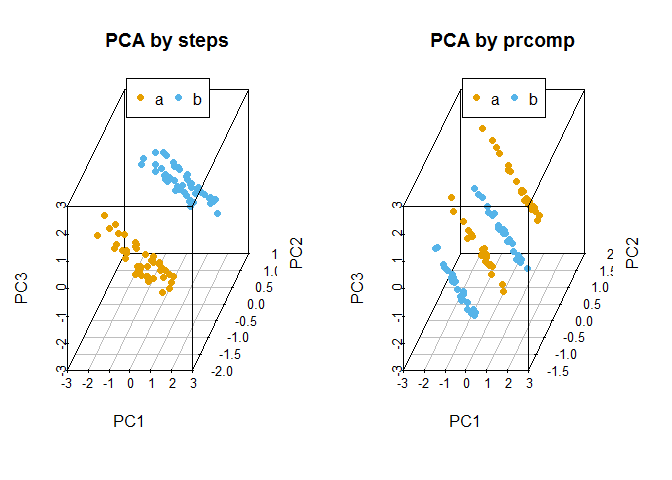
#par(mfrow=c(1,1))自定义PCA计算函数
ct_PCA <- function(data, center=TRUE, scale=TRUE){
data_norm <- scale(data, center=center, scale=scale)
data_norm_cov <- crossprod(as.matrix(data_norm)) / (nrow(data_norm)-1)
data_eigen <- eigen(data_norm_cov)
rotation <- data_eigen$vectors
label <- paste0('PC', c(1:ncol(rotation)))
colnames(rotation) <- label
sdev <- sqrt(data_eigen$values)
data_new <- data_norm %*% rotation
colnames(data_new) <- label
ct_pca <- list('rotation'=rotation, 'x'=data_new, 'sdev'=sdev)
return(ct_pca)
}比较有无scale对聚类的影响,从图中可以看到,如果不对数据进行scale处理,样品的聚类结果更像原始数据,本身数值大的基因对主成分的贡献会大。如果关注的是每个变量自身的实际方差对样品分类的贡献,则不应该SCALE;如果关注的是变量的相对大小对样品分类的贡献,则应该SCALE,以防数值高的变量导入的大方差引入的偏见。
data3_pca_noscale_step = ct_PCA(data3[,1:3], center=TRUE, scale=FALSE)
# 特征向量
data3_pca_noscale_step$rotation
## PC1 PC2 PC3
## [1,] 0.9999446062 0.010502677 -0.0006915646
## [2,] 0.0006682513 0.002223103 0.9999973056
## [3,] -0.0105041862 0.999942374 -0.0022159613
# 新变量
data3_pca_noscale_pc <- data3_pca_noscale_step$x
#library(scatterplot3d)
colorl <- c("#E69F00", "#56B4E9")
# Extract same number of colors as the Group and same Group would have same color.
colors <- colorl[as.numeric(data3$Group)]
# 1 row 2 columns
par(mfrow=c(2,2))
scatterplot3d(data3[,c(1,3,2)], color=colors, angle=55, pch=16, main="Original data")
legend("top", legend=levels(data3$Group), col=colorl, pch=16, xpd=T, horiz=T)
scatterplot3d(data3_pca_noscale_pc, color=colors,angle=55, pch=16, main="PCA (no scale)")
legend("top", legend=levels(data3$Group), col=colorl, pch=16, xpd=T, horiz=T)
scatterplot3d(data3_center_scale[,c(1,3,2)], color=colors, angle=55, pch=16,
main="Original data (scale)")
legend("top", legend=levels(data3$Group), col=colorl, pch=16, xpd=T, horiz=T)
scatterplot3d(data3_new, color=colors,angle=55, pch=16, main="PCA (scale)")
legend("top", legend=levels(data3$Group), col=colorl, pch=16, xpd=T, horiz=T)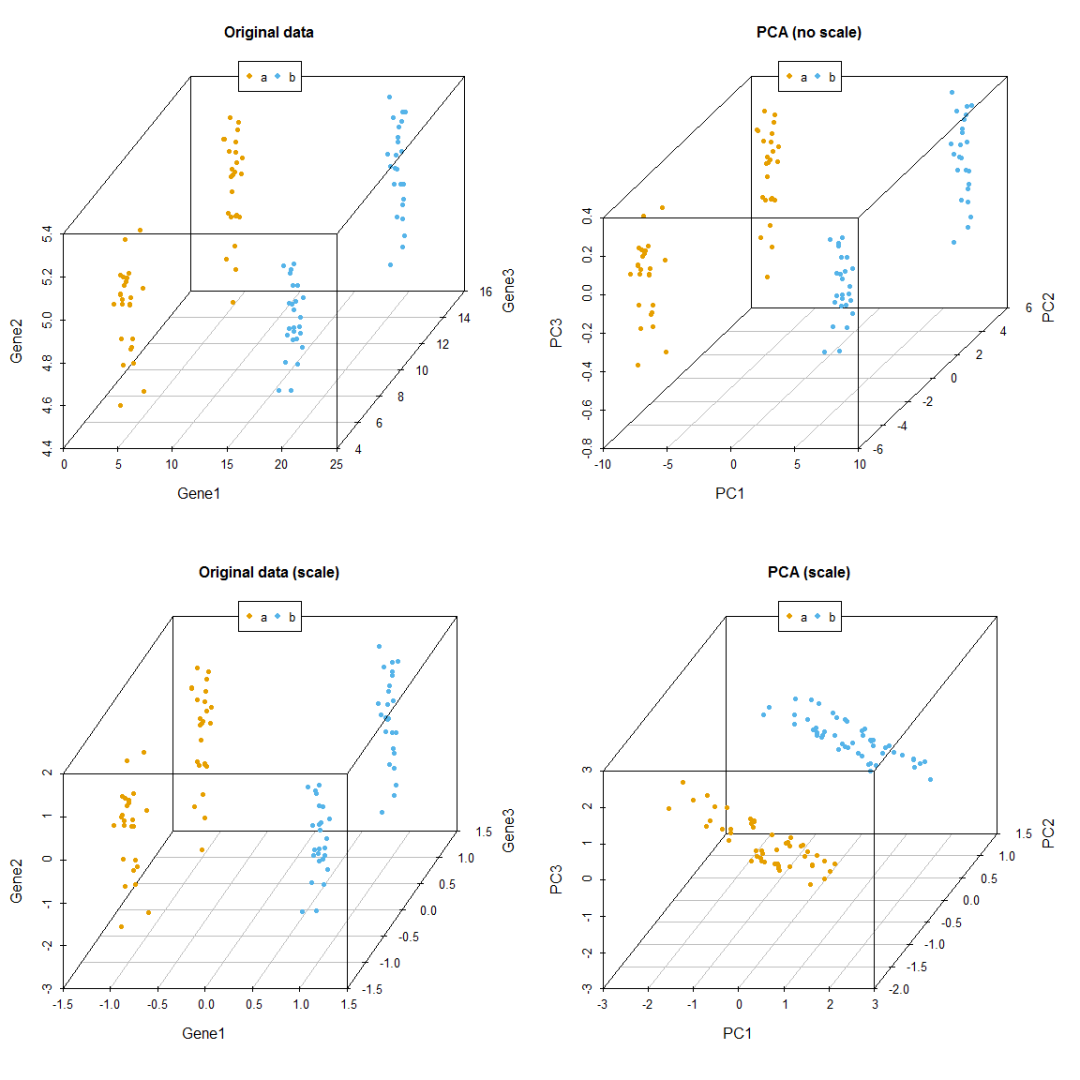
#par(mfrow=c(1,1))PCA结果解释
prcomp函数会返回主成分的标准差、特征向量和主成分构成的新矩阵。接下来,探索下不同主成分对数据差异的贡献和主成分与原始变量的关系。
主成分的平方为为特征值,其含义为每个主成分可以解释的数据差异,计算方式为
eigenvalues = (pca$sdev)^2每个主成分可以解释的数据差异的比例为
percent_var = eigenvalues*100/sum(eigenvalues)可以使用
summary(pca)获取以上两条信息。
这两个信息可以判断主成分分析的质量:
成功的降维需要保证在前几个为数不多的主成分对数据差异的解释可以达到80-90%。
指导选择主成分的数目:
选择的主成分足以解释的总方差大于80% (方差比例碎石图)
从前面的协方差矩阵可以看到,自动定标(scale)的变量的方差为1
(协方差矩阵对角线的值)。待选择的主成分应该是那些方差大于1的主成分,即其解释的方差大于原始变量(特征值碎石图,方差大于1,特征值也会大于1,反之亦然)。
鉴定核心变量和变量间的隐性关系:
原始变量与主成分的相关性
Variable correlation with PCs (var.cor) = loadings * sdev原始数据对主成分的贡献度
var.cor^2 / (total var.cor^2)
在测试数据中,scale后,三个主成分对数据差异的贡献度大都在30%左右,而未scale的数据,三个主成分对数据差异的贡献度相差很大。这是因为三个基因由于自身表达量级所引起的方差的差异导致它们各自对数据的权重差异,从而使主成分偏向于数值大的变量。
PCA应用于测试数据
前面用到一组比较大的测试数据集,并做了PCA分析,现在测试不同的处理对结果的影响。
首先回顾下我们用到的数据。
library("gridExtra")
# Pay attention to the format of PCA input
# Rows are samples and columns are variables
# data4_use_log2_t <- t(data4_use_log2)
# Add group column for plotting
# data4_use_log2_label <- as.data.frame(data4_use_log2_t)
# data4_use_log2_label$group <- group
kable(corner(data4_use_log2_label), digits=3, caption="Single cell gene expression data")| MT2A | ANXA1 | ARHGDIB | RND3 | BLVRB | |
|---|---|---|---|---|---|
| Hi_2338_1 | 12.211 | 11.837 | 9.543 | 9.762 | 9.162 |
| Hi_2338_2 | 11.306 | 8.099 | 8.072 | 10.107 | 9.423 |
| Hi_2338_3 | 11.926 | 10.689 | 9.721 | 9.388 | 9.694 |
| Hi_2338_4 | 10.974 | 9.387 | 9.883 | 8.792 | 9.767 |
| Hi_2338_5 | 14.604 | 10.375 | 9.970 | 8.815 | 9.350 |
比较对数运算和scale对样品分类的影响。
ct_pca_2d_plot <- function(pca, data_with_label, labelName='group', title='PCA') {
# sdev: standard deviation of the principle components.
# Square to get variance
percentVar <- pca$sdev^2 / sum( pca$sdev^2)
#data <- data_with_label
#data[labelName] <- factor(unlist(data[labelName]))
level <- length(unique(unlist(data_with_label[labelName])))
shapes = (1:level)%%30 # maximum allow 30 types of symbols
p = autoplot(pca, data=data_with_label, colour=labelName, shape=labelName) +
scale_shape_manual(values=shapes) +
xlab(paste0("PC1 (", round(percentVar[1]*100), "% variance)")) +
ylab(paste0("PC2 (", round(percentVar[2]*100), "% variance)")) +
theme_bw() + theme(legend.position="right") + labs(title=title) +
theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank())
return(p)
}
# By default, prcomp will centralized the data using mean.
# Normalize data for PCA by dividing each data by column standard deviation.
# Often, we would normalize data.
# Only when we care about the real number changes other than the trends,
# `scale` can be set to TRUE.
# We will show the differences of scaling and un-scaling effects.
data4_use_t <- t(data4_use)
ori_scale_pca_test <- prcomp(data4_use_t, scale=T)
ori_no_scale_pca_test <- prcomp(data4_use_t, scale=F)
log_scale_pca_test <- prcomp(data4_use_log2_t, scale=T)
log_no_scale_pca_test <- prcomp(data4_use_log2_t, scale=F)
ori_scale_pca_plot = ct_pca_2d_plot(ori_scale_pca_test, data4_use_log2_label,
title="Scaled original data")
ori_no_scale_pca_plot = ct_pca_2d_plot(ori_no_scale_pca_test, data4_use_log2_label,
title="Un-scaled original data")
log_scale_pca_plot = ct_pca_2d_plot(log_scale_pca_test, data4_use_log2_label,
title="Scaled log transformed data")
log_no_scale_pca_plot = ct_pca_2d_plot(log_no_scale_pca_test, data4_use_log2_label,
title="Un-scaled log transformed data")
a__ <- grid.arrange(ori_scale_pca_plot, ori_no_scale_pca_plot, log_scale_pca_plot,
log_no_scale_pca_plot, ncol=2)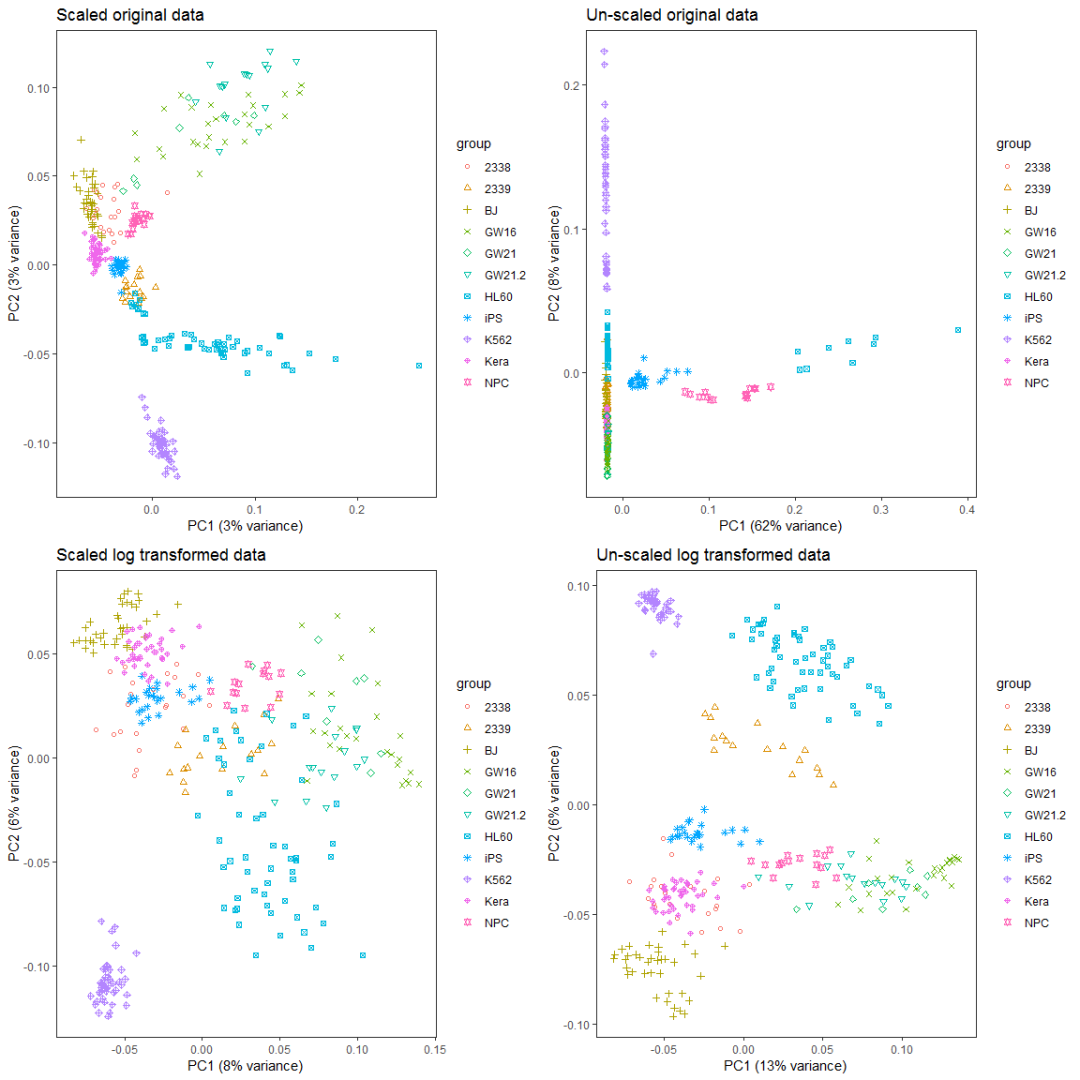
如果首先提取500个变化最大的基因,再执行PCA分析会怎样呢?
data4_use_mad <- apply(data4_use, 1, mad)
data4_use_mad_top500 <- t(data4_use[rev(order(data4_use_mad))[1:500],])
data4_use_log2_mad <- apply(data4_use_log2, 1, mad)
data4_use_log2_mad_top500 <- t(data4_use_log2[rev(order(data4_use_log2_mad))[1:500],])
ori_scale_pca_top500 <- prcomp(data4_use_mad_top500, scale=T)
ori_no_scale_pca_top500 <- prcomp(data4_use_mad_top500, scale=F)
log_scale_pca_top500 <- prcomp(data4_use_log2_mad_top500, scale=T)
log_no_scale_pca_top500 <- prcomp(data4_use_log2_mad_top500, scale=F)
ori_scale_pca_plot_t5 = ct_pca_2d_plot(ori_scale_pca_top500, data4_use_log2_label,
title="Scaled original data")
ori_no_scale_pca_plot_t5 = ct_pca_2d_plot(ori_no_scale_pca_top500,
data4_use_log2_label, title="Un-scaled original data")
log_scale_pca_plot_t5 = ct_pca_2d_plot(log_scale_pca_top500,
data4_use_log2_label, title="Scaled log transformed data")
log_no_scale_pca_plot_t5 = ct_pca_2d_plot(log_no_scale_pca_top500,
data4_use_log2_label, title="Un-scaled log transformed data")
a__ <- grid.arrange(ori_scale_pca_plot_t5, ori_no_scale_pca_plot_t5,
log_scale_pca_plot_t5, log_no_scale_pca_plot_t5, ncol=2)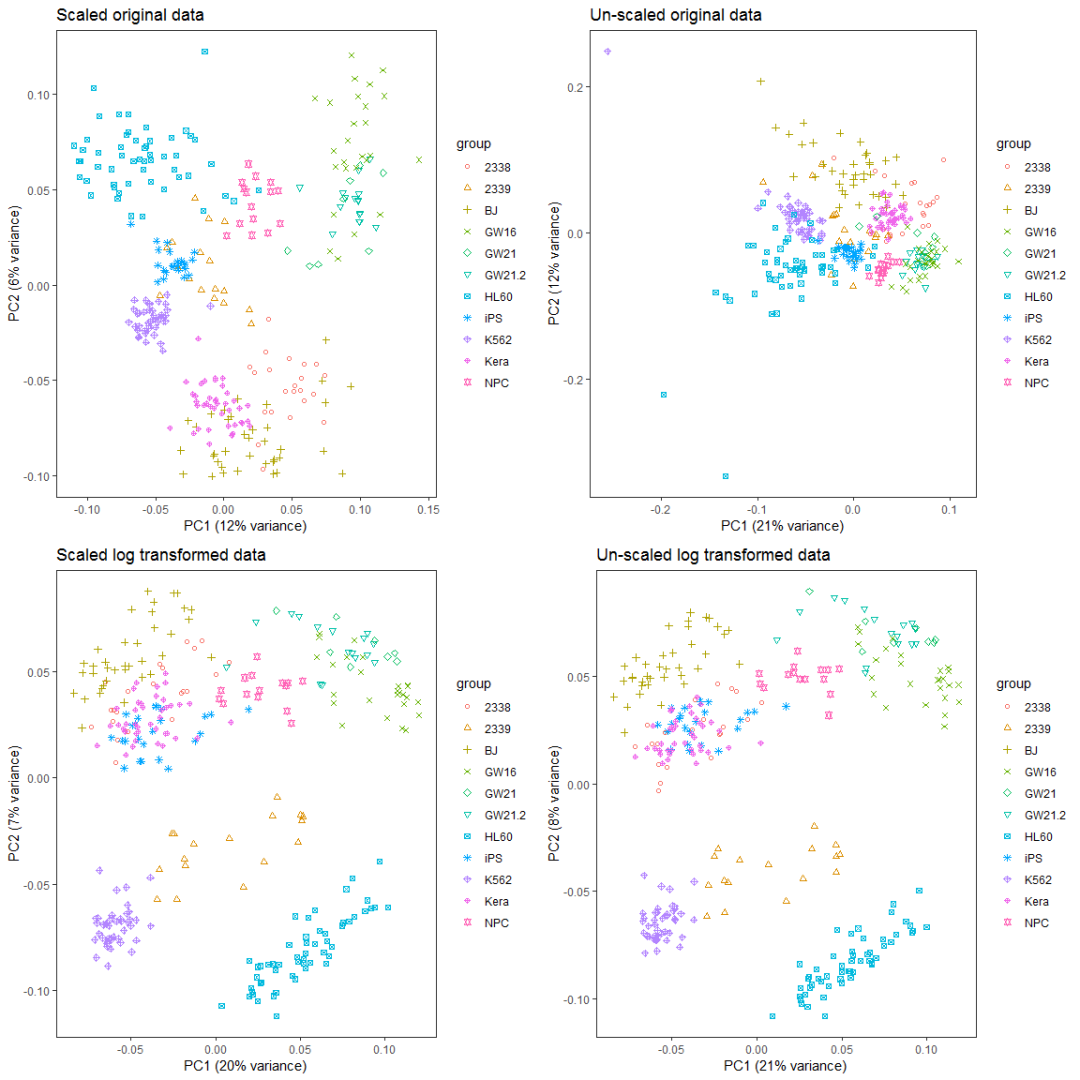
### PCA注意事项
一般说来,在PCA之前原始数据需要中心化(centering,数值减去平均值)。中心化的方法很多,除了平均值中心化(mean-centering)外,还包括其它更稳健的方法,比如中位数中心化等。
除了中心化以外,定标 (Scale, 数值除以标准差)
也是数据前处理中需要考虑的一点。如果数据没有定标,则原始数据中方差大的变量对主成分的贡献会很大。数据的方差与其量级成指数关系,比如一组数据(1,2,3,4)的方差是1.67,而(10,20,30,40)的方差就是167,数据变大10倍,方差放大了100倍。但是定标(scale)可能会有一些负面效果,因为定标后变量之间的权重就是变得相同。如果我们的变量中有噪音的话,我们就在无形中把噪音和信息的权重变得相同,但PCA本身无法区分信号和噪音。在这样的情形下,我们就不必做定标。
一般而言,对于度量单位不同的指标或是取值范围彼此差异非常大的指标不直接由其协方差矩阵出发进行主成分分析,而应该考虑对数据的标准化。比如度量单位不同,有万人、万吨、万元、亿元,而数据间的差异性也非常大,小则几十大则几万,因此在用协方差矩阵求解主成分时存在协方差矩阵中数据的差异性很大。在后面提取主成分时发现,只提取了一个主成分,而此时并不能将所有的变量都解释到,这就没有真正起到降维的作用。此时就需要对数据进行定标(scale),这样提取的主成分可以覆盖更多的变量,这就实现主成分分析的最终目的。但是对原始数据进行标准化后更倾向于使得各个指标的作用在主成分分析构成中相等。对于数据取值范围不大或是度量单位相同的指标进行标准化处理后,其主成分分析的结果与仍由协方差矩阵出发求得的结果有较大区别。这是因为对数据标准化的过程实际上就是抹杀原有变量离散程度差异的过程,标准化后方差均为1,而实际上方差是对数据信息的重要概括形式,也就是说,对原始数据进行标准化后抹杀了一部分重要信息,因此才使得标准化后各变量在主成分构成中的作用趋于相等。因此,对同度量或是取值范围在同量级的数据还是直接使用非定标数据求解主成分为宜。
中心化和定标都会受数据中离群值(outliers)或者数据不均匀(比如数据被分为若干个小组)的影响,应该用更稳健的中心化和定标方法。
PCA也存在一些限制,例如它可以很好的解除线性相关,但是对于高阶相关性就没有办法了,对于存在高阶相关性的数据,可以考虑Kernel
PCA,通过Kernel函数将非线性相关转为线性相关,关于这点就不展开讨论了。另外,PCA假设数据各主特征是分布在正交方向上,如果在非正交方向上存在几个方差较大的方向,PCA的效果就大打折扣了。
参考资料
https://www.zhihu.com/question/20998460
PCA
教程1PCA 文字化描述
pca1
ggplot2
axisscatterplot3D
稳健PCA
http://www.nlpca.org/pca_principal_component_analysis.html
Data
centeringSample R
markdown矩阵特征值,对称矩阵的对角化
Detail usage and visualization of prcomp
resultggplot2 side by side
plot
这篇Nature子刊文章的蛋白组学数据PCA分析竟花费了我两天时间来重现|附全过程代码
复现nature communication PCA原图|代码分析(一)
本文节选自:这个为生信学习和生信作图打造的开源R教程真香!!!