让你的单细胞数据动起来!|iCellR(一)
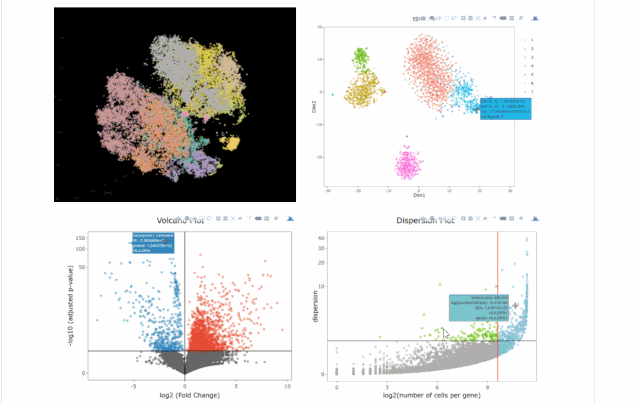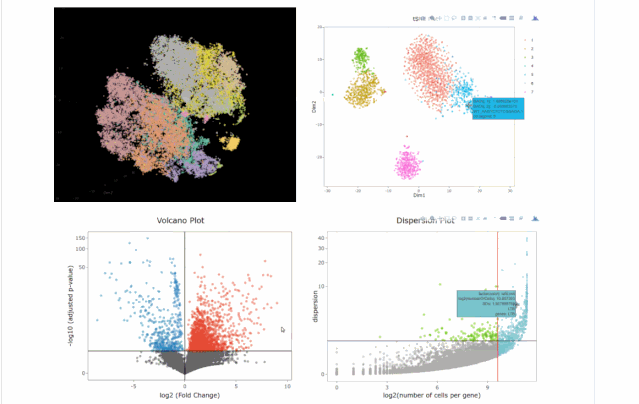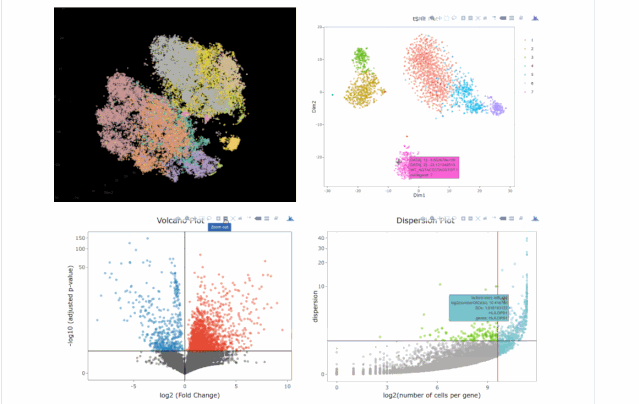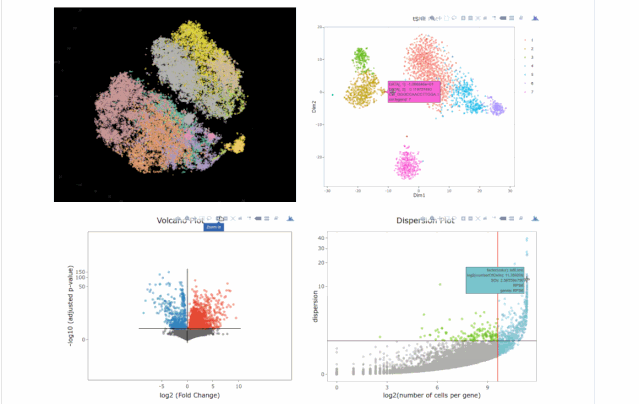
今天在翻阅single cell 的github时候,我看见了这个R包,允许我们处理各种来自单细胞测序技术的数据,如scRNA-seq,scVDJ-seq和CITE-Seq。
想看整套的学习流程还可以戳这里:
https://vimeo.com/337822487
iCellR优点
Single(i)Cell R软件包(iCellR)在分析pipeline的每个步骤提供前所未有的灵活性,包括标准化,聚类,降维,插补,可视化等。可以通过无监督和有监督聚类进行分析, 此外,该工具包提供2D和3D交互式可视化(一个震撼的交互型3D可视化R包 - 可直接转ggplot2图为3D),差异表达分析,基于细胞、基因和聚类的filter,数据合并,dropouts的标准化,数据插补方法,校正批次差异,找到标记基因的工具聚类和条件,预测细胞类型和伪时序分析(NBT|45种单细胞轨迹推断方法比较,110个实际数据集和229个合成数据集)。感觉能做的真的好多啊!
安装iCellR
# 从镜像中进行安装
install.packages("iCellR")
# 也可以从github中进行安装
#library(devtools)
#install_github("rezakj/iCellR")
# or
#git clone https://github.com/rezakj/iCellR.git
#R
#install.packages('iCellR/', repos = NULL, type="source")
下载演示数据
ion(samples = c("sample1","sample2","sample3"),
# 设置需要下载的文件夹位置
setwd("/your/download/directory")
# 通过URL进行数据下载
sample.file.url = "https://s3-us-west-2.amazonaws.com/10x.files/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz"
# 下载文件
download.file(url = sample.file.url,
destfile = "pbmc3k_filtered_gene_bc_matrices.tar.gz",
method = "auto")
# 解压缩.
untar("pbmc3k_filtered_gene_bc_matrices.tar.gz")更多的数据可以通过这里获得:
https://genome.med.nyu.edu/results/external/iCellR/
如何使用iCellR分析scRNA-seq数据
1. 加载iCellR包和下载的PBMC样本数据。
library("iCellR")
my.data <- load10x("filtered_gene_bc_matrices/hg19/")
#该目录有barcodes.tsv,genes.tsv和matrix.mtx文件,将10x的数据读入数据框
# 如果是SMART-seq2中有名的小鼠全器官数据,读入方式如下
# my.data = read.delim("FACS/Kidney-counts.csv", sep=",", header=TRUE)2. 汇总数据
dim(my.data)
# [1] 32738 2700
# 将示例数据进行拆分为3个samples
sample1 <- my.data[1:900]
sample2 <- my.data[901:1800]
sample3 <- my.data[1801:2700]
# 合并示例文件my.data <- data.aggregat condition.names = c("WT","KO","Ctrl"))
# 查看数据
head(my.data)[1:2]
# WT_AAACATACAACCAC-1 WT_AAACATTGAGCTAC-1
#A1BG 0 0
#A1BG.AS1 0 0
#A1CF 0 0
#A2M 0 0
#A2M.AS1 0 03. 制作iCellR类的对象。
my.obj <- make.obj(my.data)
my.obj
###################################
,--. ,-----. ,--.,--.,------.
`--'' .--./ ,---. | || || .--. '
,--.| | | .-. :| || || '--'.'
| |' '--'\ --. | || || |
`--' `-----' `----'`--'`--'`--' '--'
###################################
An object of class iCellR version: 0.99.0
Raw/original data dimentions (rows,columns): 32738,2700
Data conditions in raw data: Ctrl,KO,WT (900,900,900)
Row names: A1BG,A1BG.AS1,A1CF ...
Columns names: WT_AAACATACAACCAC.1,WT_AAACATTGAGCTAC.1,WT_AAACATTGATCAGC.1 ...
###################################
QC stats performed: FALSE , PCA performed: FALSE , CCA performed: FALSE
Clustering performed: FALSE , Number of clusters: 0
tSNE performed: FALSE , UMAP performed: FALSE , DiffMap performed: FALSE
Main data dimentions (rows,columns): 0 0
Normalization factors: ...
Imputed data dimentions (rows,columns): 0 0
############## scVDJ-Seq ###########
VDJ data dimentions (rows,columns): 0 0
############## CITE-Seq ############
ADT raw data dimentions (rows,columns): 0 0
ADT main data dimentions (rows,columns): 0 0
ADT columns names: ...
ADT row names: ...
######## iCellR object made ########4. 进行质控(QC)
my.obj <- qc.stats(my.obj) # 计算每个细胞的UMI数和基因数,还有细胞周期基因和线粒体基因('mito.percent')的百分比5. 细胞周期预测
和上一个推送的细胞周期预测方法还是较为一致的,都是通过评分进行周期的预测,
my.obj <- cc(my.obj, s.genes = s.phase, g2m.genes = g2m.phase)
head(my.obj@stats)
# CellIds nGenes UMIs mito.percent
#WT_AAACATACAACCAC.1 WT_AAACATACAACCAC.1 781 2421 0.030152829
#WT_AAACATTGAGCTAC.1 WT_AAACATTGAGCTAC.1 1352 4903 0.037935958
#WT_AAACATTGATCAGC.1 WT_AAACATTGATCAGC.1 1131 3149 0.008891712
#WT_AAACCGTGCTTCCG.1 WT_AAACCGTGCTTCCG.1 960 2639 0.017430845
#WT_AAACCGTGTATGCG.1 WT_AAACCGTGTATGCG.1 522 981 0.012232416
#WT_AAACGCACTGGTAC.1 WT_AAACGCACTGGTAC.1 782 2164 0.016635860
# S.phase.probability g2m.phase.probability S.Score
#WT_AAACATACAACCAC.1 0.0012391574 0.0004130525 0.030569081
#WT_AAACATTGAGCTAC.1 0.0002039568 0.0004079135 -0.077860621
#WT_AAACATTGATCAGC.1 0.0003175611 0.0019053668 -0.028560560
#WT_AAACCGTGCTTCCG.1 0.0007578628 0.0011367942 0.001917225
#WT_AAACCGTGTATGCG.1 0.0000000000 0.0020387360 -0.020085210
#WT_AAACGCACTGGTAC.1 0.0000000000 0.0000000000 -0.038953135
# G2M.Score Phase
#WT_AAACATACAACCAC.1 -0.0652390011 S
#WT_AAACATTGAGCTAC.1 -0.1277015099 G1
#WT_AAACATTGATCAGC.1 -0.0036505733 G1
#WT_AAACCGTGCTTCCG.1 -0.0499511543 S
#WT_AAACCGTGTATGCG.1 0.0009426363 G2M
#WT_AAACGCACTGGTAC.1 -0.0680240629 G1
# 绘制细胞周期图
pie(table(my.obj@stats$Phase))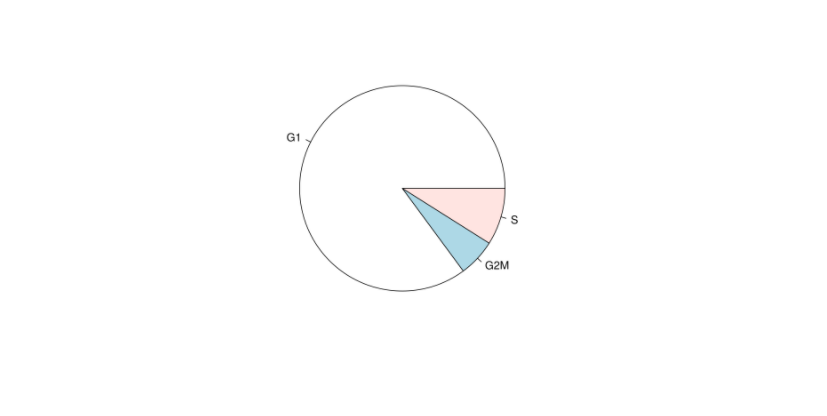
6. Plot QC
默认情况下的绘图功能都将创建交互式html文件,可以设置interactive = FALSE来关闭。
# 绘制 三个分组中UMIs, genes和 percent mito的beanplot分布图,想单独画地话可以用`?stats.plot`help一下如何修改参数
stats.plot(my.obj,
plot.type = "all.in.one",
out.name = "UMI-plot",
interactive = FALSE,
cell.color = "slategray3",
cell.size = 1,
cell.transparency = 0.5,
box.color = "red",
box.line.col = "green")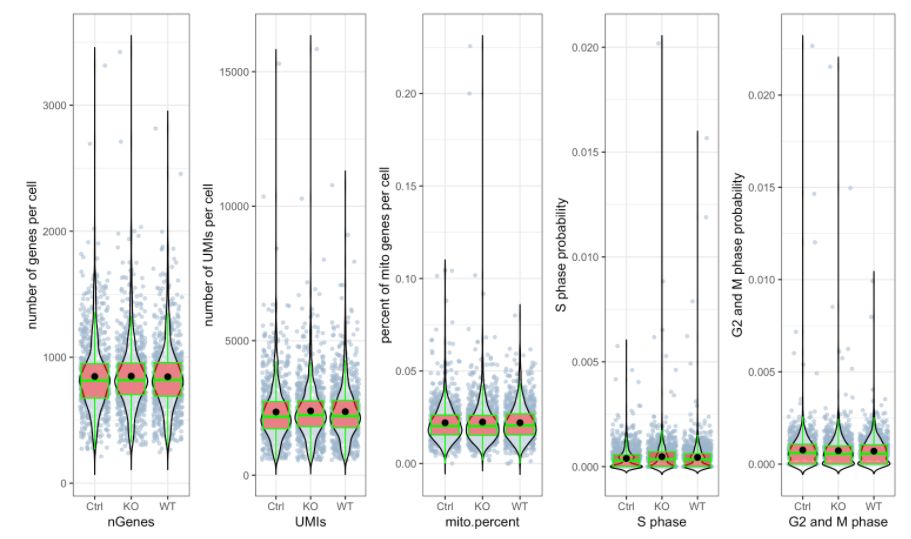
# 绘制散点图查看线粒体基因和基因分布,注意参数`plot.type`
stats.plot(my.obj, plot.type = "point.mito.umi", out.name = "mito-umi-plot")###该图为线粒体基因和UMI的相关表达分布(一般原则下会认为线粒体基因比例较高可能为破碎衰老细胞,可以设置cutoff值去掉,但如果研究为高代谢或本身耗能较多的细胞时需要考虑)
stats.plot(my.obj, plot.type = "point.gene.umi", out.name = "gene-umi-plot")###此图为UMI和GENE的表达分布(原则上遵循线性分布)
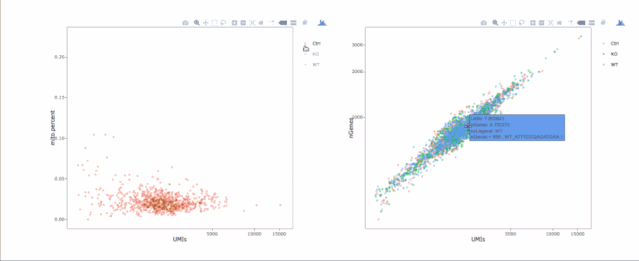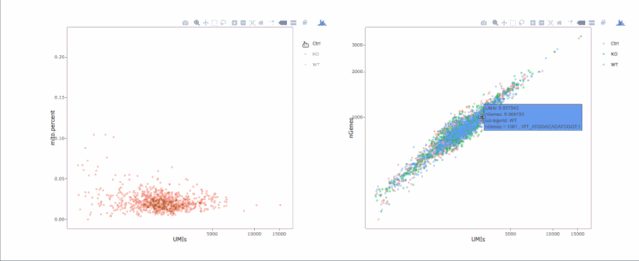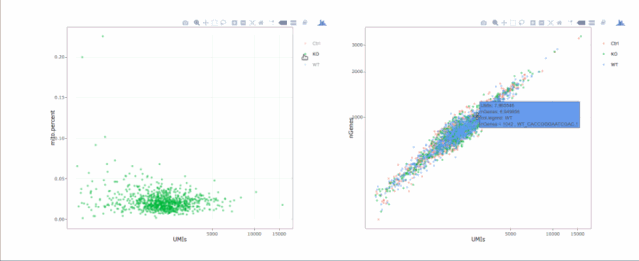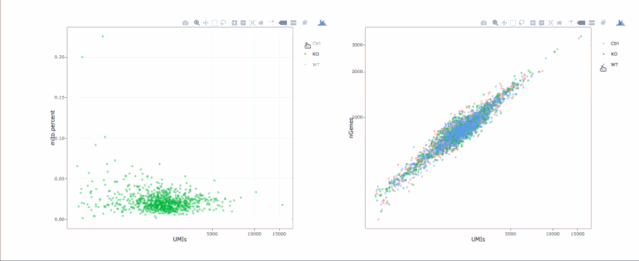
7. 过滤细胞
my.obj <- cell.filter(my.obj,
min.mito = 0,
max.mito = 0.05,
min.genes = 200,
max.genes = 2400,
min.umis = 0,
max.umis = Inf)
#[1] "cells with min mito ratio of 0 and max mito ratio of 0.05 were filtered." 保留线粒体基因比例在0到0.05的细胞
#[1] "cells with min genes of 200 and max genes of 2400 were filtered." 保留基因数在200到2400之间的细胞
#[1] "No UMI number filter" 不通过UMI数和细胞ID来筛选
#[1] "No cell filter by provided gene/genes"
#[1] "No cell id filter"
#[1] "filters_set.txt file has beed generated and includes the filters set for this experiment."筛选之后会生成一个文件filter_set,含有实验的筛选设置
# more examples 也可以通过基因和细胞ID来筛选
# my.obj <- cell.filter(my.obj, filter.by.gene = c("RPL13","RPL10")) # filter our cell having no counts for these genes
# my.obj <- cell.filter(my.obj, filter.by.cell.id = c("WT_AAACATACAACCAC.1")) # filter our cell cell by their cell ids.
# 查看剩下的细胞数(线粒体基因比例在0到0.05,基因数在200到2400之间)
dim(my.obj@main.data)
#[1] 32738 2637
# 过滤掉了63个细胞
8. 进行标准化
可以根据自己的研究对数据进行标准化。还可以使用iCellR以外的工具对数据进行标准化,并将数据导入iCellR。对于大多数单细胞研究,建议使用“ranked.glsf”标准化。这种标准化化比较适合具有大量零的矩阵。且在合并数据(操作参考步骤2:汇总数据)的情况下,也能对批次进行校正。
my.obj <- norm.data(my.obj,
norm.method = "ranked.glsf",
top.rank = 500) # best for scRNA-Seq
# **更多的标准化方式**
#my.obj <- norm.data(my.obj, norm.method = "ranked.deseq", top.rank = 500)
#my.obj <- norm.data(my.obj, norm.method = "deseq") # best for bulk RNA-Seq
#my.obj <- norm.data(my.obj, norm.method = "global.glsf") # best for bulk RNA-Seq
#my.obj <- norm.data(my.obj, norm.method = "rpm", rpm.factor = 100000) # best for bulk RNA-Seq
#my.obj <- norm.data(my.obj, norm.method = "spike.in", spike.in.factors = NULL)
#my.obj <- norm.data(my.obj, norm.method = "no.norm") # if the data is already normalized
9. 进行二次QC
my.obj <- qc.stats(my.obj,which.data = "main.data")
### 该次QC的主要目的就是计算UMI数量,每个细胞的基因以及每个细胞和细胞周期基因的线粒体基因的百分比。从图中可以看出其细胞周期相关基因表达较低,线粒体基因表达比例也较低
stats.plot(my.obj,
plot.type = "all.in.one",
out.name = "UMI-plot",
interactive = F,
cell.color = "slategray3",
cell.size = 1,
cell.transparency = 0.5,
box.color = "red",
box.line.col = "green",
back.col = "white")
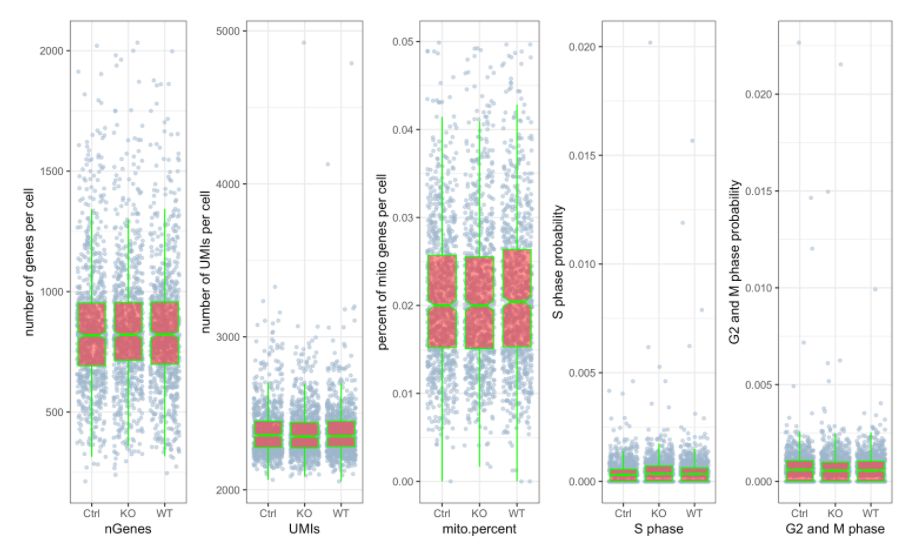
10. Scale data
my.obj <- data.scale(my.obj) # 对数据进行中心化和标准化处理11.进行gene统计
my.obj <- gene.stats(my.obj, which.data = "main.data")
head(my.obj@gene.data[order(my.obj@gene.data$numberOfCells, decreasing = T),]) # 将数据通过拥有该基因的细胞数按从大到小进行排序
# genes numberOfCells totalNumberOfCells percentOfCells meanExp
#30303 TMSB4X 2637 2637 100.00000 38.55948
#3633 B2M 2636 2637 99.96208 45.07327
#14403 MALAT1 2636 2637 99.96208 70.95452
#27191 RPL13A 2635 2637 99.92416 32.29009
#27185 RPL10 2632 2637 99.81039 35.43002
#27190 RPL13 2630 2637 99.73455 32.32106
# SDs condition
#30303 7.545968e-15 all
#3633 2.893940e+01 all
#14403 7.996407e+01 all
#27191 2.783799e+01 all
#27185 2.599067e+01 all
#27190 2.661361e+01 all12.制作聚类的基因模型
在bulk RNA-seq数据中,用平均表达值meanExp排名前500的基因(设置参数base.mean.rank对样本进行聚类是非常常见的,主要原因是为了降低噪声。在scRNA-seq数据中也可以这样做。并加上ranked.glsf归一化,会较为擅长处理具有大量零的矩阵。
# 看一下高变基因集
make.gene.model(my.obj, my.out.put = "plot",
dispersion.limit = 1.5,
base.mean.rank = 500,
no.mito.model = T,
mark.mito = T,
interactive = F,
no.cell.cycle = T,
out.name = "gene.model")
# 将高变基因集注入`my.obj` 中
my.obj <- make.gene.model(my.obj, my.out.put = "data",
dispersion.limit = 1.5,
base.mean.rank = 500,
no.mito.model = T,
mark.mito = T,
interactive = F,
no.cell.cycle = T,
out.name = "gene.model")
head(my.obj@gene.model) # 高变基因
# "ACTB" "ACTG1" "ACTR3" "AES" "AIF1" "ALDOA"
# get html plot (optional)
#make.gene.model(my.obj, my.out.put = "plot",
# dispersion.limit = 1.5,
# base.mean.rank = 500,
# no.mito.model = T,
# mark.mito = T,
# interactive = T,
# out.name = "plot4_gene.model")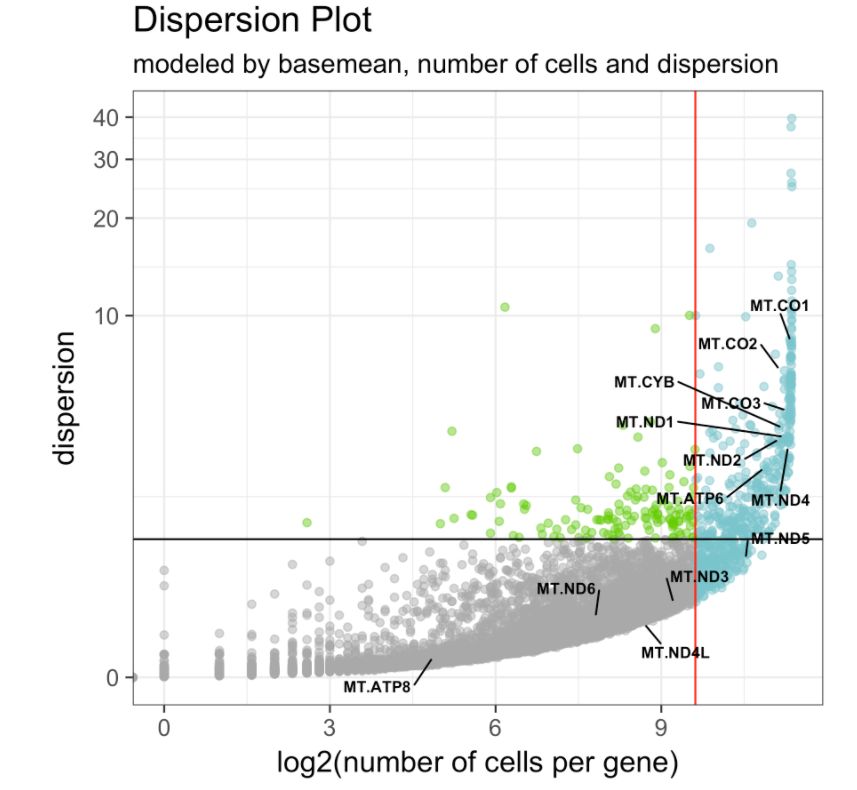
13. 进行降维
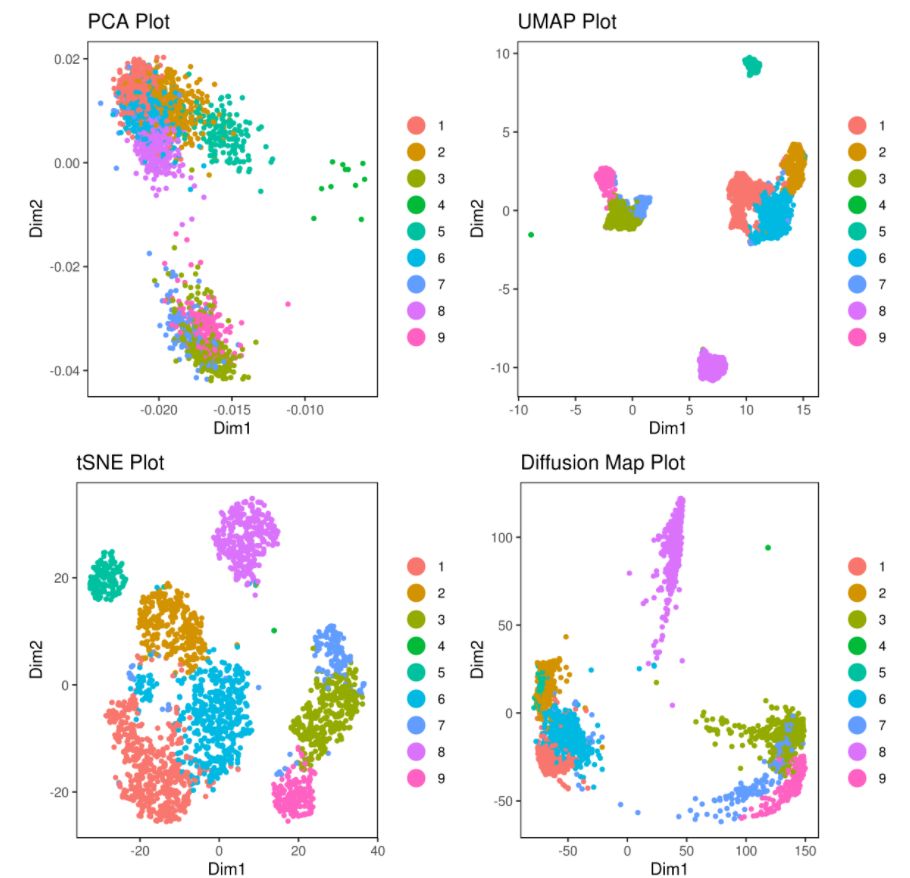
降维是为了在信息损失较少的情况下,以比原始特征数量少得多的特征(基因)来表示数据,更好地捕捉细胞之间的关系,常见降维的方法有PCA,t-SNE,umap和diffusion map。(Hemberg-lab单细胞转录组数据分析(十一)- Scater单细胞表达谱PCA可视化)
PCA分析
my.obj <- run.pca(my.obj, method = "gene.model", gene.list = my.obj@gene.model,data.type = "main",batch.norm = F)
opt.pcs.plot(my.obj)
##下图为典型的滚石图,当趋势越趋于平缓表明其PC值作用越小,下图PCA取到10。这一步是可选的,可能在一些情况下也是不适用的。# 2 round PCA (to find top genes in the first 10 PCs and re-run PCA for better clustering
## This is optional and might not be good in some cases
length(my.obj@gene.model)
# 585
my.obj <- find.dim.genes(my.obj, dims = 1:10,top.pos = 20, top.neg = 20) # (optional)
length(my.obj@gene.model)
# 208
# second round PC
my.obj <- run.pca(my.obj, method = "gene.model", gene.list = my.obj@gene.model,data.type = "main",batch.norm = F)
my.obj@opt.pcs

Hemberg-lab单细胞转录组数据分析(十一)- Scater单细胞表达谱PCA可视化
更多降维方法
# tSNE
my.obj <- run.pc.tsne(my.obj, dims = 1:10)
# UMAP
my.obj <- run.umap(my.obj, dims = 1:10, method = "naive")
# or
# my.obj <- run.umap(my.obj, dims = 1:10, method = "umap-learn")
# diffusion map
# this requires python packge phate
# pip install --user phate
# Install phateR version 2.9
# wget https://cran.r-project.org/src/contrib/Archive/phateR/phateR_0.2.9.tar.gz
# install.packages('phateR/', repos = NULL, type="source")
library(phateR)
my.obj <- run.diffusion.map(my.obj, dims = 1:10, method = "phate")
14. 对细胞进行聚类分析
“聚类是针对给定的样本,依据它们特征的相似度或距离,将其归并到若干个“类”或“簇”的数据分析问题。一个类是样本的一个子集。直观上,相似的样本聚集在相同的类,不相似的样本分散在不同的类。这里,样本之间的相似度或距离起着重要作用。”
降维是对feature (gene) 进行操作,而聚类是对sample进行操作,建议在得到高变基因以及降维后操作。(基因表达聚类分析之初探SOM)
建议使用以下默认选项:
my.obj <- run.clustering(my.obj,
clust.method = "kmeans", # 聚类方式为k-means
dist.method = "euclidean", # 距离计算是欧氏距离,有非常明确的空间距离概念
index.method = "silhouette",
max.clust = 25,
min.clust = 2,
dims = 1:10)
# 如果要手动设置cluster数量,而不使用预测的最佳数量,请将最小值和最大值设置为所需的数量:
#my.obj <- run.clustering(my.obj,
# clust.method = "ward.D",
# dist.method = "euclidean",
# index.method = "ccc",
# max.clust = 8,
# min.clust = 8,
# dims = 1:10)
# more examples
#my.obj <- run.clustering(my.obj,
# clust.method = "ward.D",
# dist.method = "euclidean",
# index.method = "kl",
# max.clust = 25,
# min.clust = 2,
# dims = 1:10)
15. 数据可视化
# clusters
A= cluster.plot(my.obj,plot.type = "pca",interactive = F)
B= cluster.plot(my.obj,plot.type = "umap",interactive = F)
C= cluster.plot(my.obj,plot.type = "tsne",interactive = F)
D= cluster.plot(my.obj,plot.type = "diffusion",interactive = F)
#下图通过不同降维方式进行展示,其实我们可以看出,在该组数据中还是t-sne降维后的可视化表达方式最好(细胞之间的分离度较好)
library(gridExtra)
grid.arrange(A,B,C,D)
# conditions
A= cluster.plot(my.obj,plot.type = "pca",col.by = "conditions",interactive = F)
B= cluster.plot(my.obj,plot.type = "umap",col.by = "conditions",interactive = F)
C= cluster.plot(my.obj,plot.type = "tsne",col.by = "conditions",interactive = F)
D= cluster.plot(my.obj,plot.type = "diffusion",col.by = "conditions",interactive = F)
#在下图中我们可以发现无论哪种降维方式均没有batch effect不同条件下的细胞重合度较好;library(gridExtra)
grid.arrange(A,B,C,D)

16. 三维图,密度图和交互式图
# 2D展示
cluster.plot(my.obj,
cell.size = 1,
plot.type = "tsne",
cell.color = "black",
back.col = "white",
col.by = "clusters",
cell.transparency = 0.5,
clust.dim = 2,
interactive = F)
# 可交互的2D图
cluster.plot(my.obj,
plot.type = "tsne",
col.by = "clusters",
clust.dim = 2,
interactive = T,
out.name = "tSNE_2D_clusters")
# 可交互的3D图
cluster.plot(my.obj,
plot.type = "tsne",
col.by = "clusters",
clust.dim = 3,
interactive = T,
out.name = "tSNE_3D_clusters")
# cluster的密度图
cluster.plot(my.obj,
plot.type = "pca",
col.by = "clusters",
interactive = F,
density=T)
# Density plot for conditions
cluster.plot(my.obj,
plot.type = "pca",
col.by = "conditions",
interactive = F,
density=T)
#下图为不同可视化方式,尽管方法多种多样,但个人感觉还是2D的方式展示是最为合适的。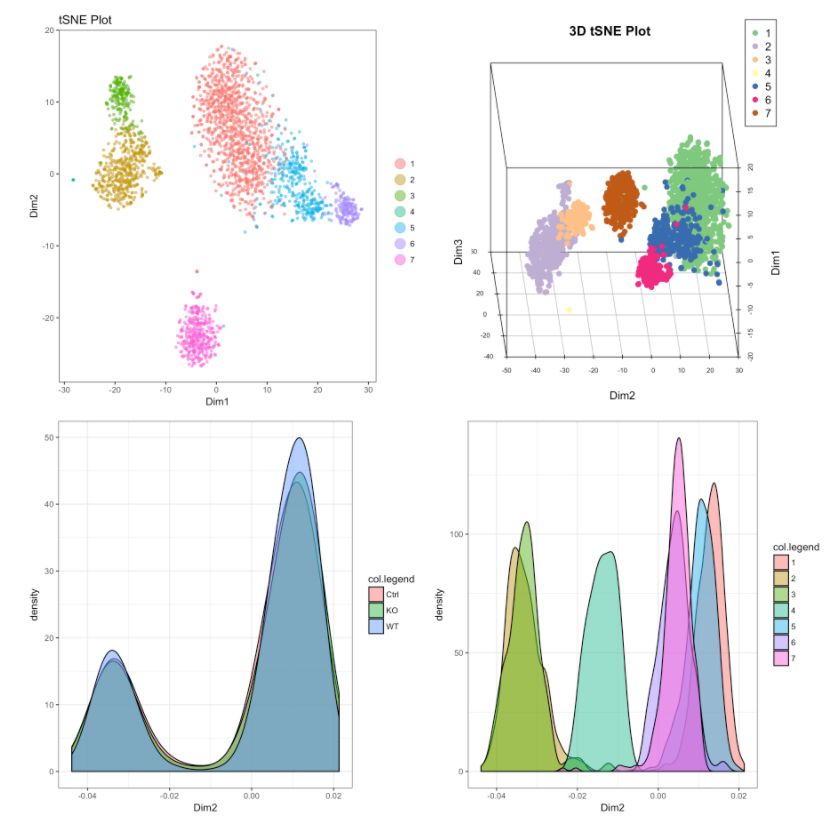
17. 不同cluster和条件下细胞比例
#如果normalize.ncell = TRUE,它将在三种状态(conditions)随机**降采样(down sample)**,因此所有条件都具有相同数量的细胞,如果为FALSE则输出原始细胞数。#下图分别是在不同条件下细胞的比例关系,KO,WT,ctrl都是我们较为常见的细胞分组。# bar plot
clust.cond.info(my.obj, plot.type = "bar", normalize.ncell = FALSE, my.out.put = "plot")
# Pie chart
clust.cond.info(my.obj, plot.type = "pie", normalize.ncell = FALSE, ,my.out.put = "plot")
# data
my.obj <- clust.cond.info(my.obj, plot.type = "bar", normalize.ncell = F)
#head(my.obj@my.freq)
# conditions clusters Freq
#1 ctrl 1 199
#2 KO 1 170
#3 WT 1 182
#4 ctrl 2 106
#5 KO 2 116
#6 WT 2 113
到这里,我们其实已经表现了许多的plot,下期接着介绍吧!
单细胞
高颜值免费在线绘图
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集