为什么说FPKM和RPKM都错了?
生物信息学习的正确姿势
NGS系列文章包括NGS基础、高颜值在线绘图和分析、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程)、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step))、批次效应处理等内容。

全文5,314字,阅读16分钟。
这本是三年多之前我发在公众号上的一篇旧文,一些偶然的机会,发现不少朋友也在讨论这个问题,因此我重新做了梳理并发出来。这样的讨论是有益的,放在今天也不过时,我也愿意和更多的朋友一起进一步讨论这个问题。
曾经(2015年),我接触了一个RNA-seq的项目,做完之后,我重新思考了FPKM和RPKM的计算,觉得它们很可能是不对的(当时是第一次接触RNA-seq数据,还不知道TPM的存在),后来查阅了一些文献终于验证了我的想法。现在我重新将这个过程记录下来。
FPKM和RPKM分别是什么
RPKM是Reads Per Kilobase per Million的缩写,它的计算方程非常简单:
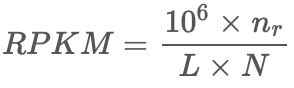
其中,
n_r 是比对至目标基因的read数量;
L 是目标基因的外显子长度之和除以1000。
因此,要注意这里的L单位是kb,不是bp;N 是总有效比对至基因组的read数量。
FPKM是Fregments Per Kilobase per Million的缩写,它的计算与RPKM极为类似,如下:
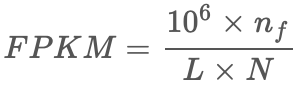
其中,n_f是比对至目标基因的fregment数量。
与RPKM唯一的区别为:F是fragments,R是reads,如果是PE(Pair-end)测序,每个fragments会有两个reads,FPKM只计算两个reads能比对到同一个转录本的fragments数量,而RPKM计算的是可以比对到转录本的reads数量而不管PE的两个reads是否能比对到同一个转录本上。如果是SE(single-end)测序,那么FPKM和RPKM计算的结果将是一致的。
以上是这两个量的计算方式,它们这样计算的目是为了解决计算RNA-seq转录本丰度时的两个bias:
相同表达丰度的转录本,往往会由于其基因长度上的差异,导致测序获得的Read(Fregment)数不同。总的来说,越长的转录本,测得的Read(Fregment)数越多;
由测序文库的不同大小而引来的差异。即同一个转录本,其测序深度越深,通过测序获得的Read(Fregment)数就越多。
FPKM和RPKM通过同时除以L(转录本长度)和N(有效比对的Read(Fregment)总数)的方式,最终将不同样本(或者同个样本在不同条件下)的转录本丰度归一化到一个能够进行量化比较的标准上。
上面的式子看起来似乎合情合理,但是它们却都做错了!
为什么FPKM/RPKM是错的
要回答这个问题,我们需要先撇开所有形式上的计算,重新思考到底什么是RNA转录本的表达丰度这个问题。
事实上,对于任何一个制备好测序文库的待测样本,它上面任何一个基因的表达量(或者说丰度),都将已是一个客观存在的值,这个值是不管你改变了多少测序环境都不会变的。而且总共有多少个不同的基因在这这一刻进行了表达,实际上也已经是客观定好了——难道不是吗?!
当我们开始以这样一种“先知”的形式来理解问题时,有趣的事情就开始出现了。
此刻,我们可以假定,对于样本X,其有一个基因g被转录了mRNA_g次,同时样本X中所有基因的转录总次数假定是mRNA_total, 那么正确描述基因g转录丰度的值应该是:
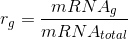
同时,样本X中其他基因的转录丰度的计算也应该和上述公式类似。除了要把分子mRNA_g换为其他基因对应的转录次数之外,分母mRNAtotal都一样。于是有趣的事情就是,所有基因转录本丰度的均值r_mean将是一个恒定不变的数,由以上定义这个数就是:
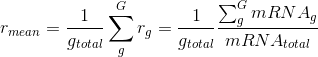
而
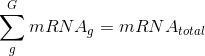
所以
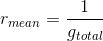
这个期望值竟然和测序状态无关!仅仅由样本中基因的总数所决定的。也就是说,对于同一个物种,不管它的样本是哪种组织(正常的或病变的),也不管有多少个不同的样本,只要它们都拥有相同数量的基因,那么它们的r_mean都将是一致的。这是一个在进行比较分析的时候,非常有意义的恒等关系。
但在实际的操作中,我们是无法直接计算这些r值的。好在只要能够保证模型本身的自洽性,我们是能通过自建一些统计量来对r值进行间接描述的。比如FPKM和RPKM,实际上它们的目的就是为了描述r。
但这里的大前提就是,所有这些要用来描述转录本丰度的统计量,都应该能等价表示出这一恒等关系。即,不管如何,这些统计量所描述出来的转录本丰度应该得是其真实丰度(r_g)的m倍(m必须是一个根据模型定出的不变值),均值也将是r_mean的m倍,只有这样才是得出有意义的结果。
现在,我们回过头来看看FPKM和RPKM的计算式,就会发现它们根本做不到!

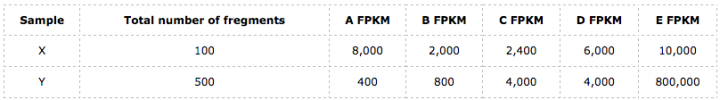
先举个例子来说明(以FPKM的计算为例),我们假定有两个来自同一个个体不同组织的样本X和Y,这个个体只有5个基因,分别为A、B、C、D和E它们的长度分别如下:

由此,我们可以得到,样本X和Y的转录本的不变量,r_mean值是:
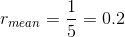
如果FPKM或RPKM是一个合适的统计量的话,那么,样本X和Y的平均FPKM(或RPKM)应该相等。
我们以FPKM的计算的为例子,以下这个表格列出的分别是样本X和Y在这5个基因分别比对上的fregment数和各自总的fregment数量:

于是,按照以上公式我们可以得到样本X和Y在这5个基因上的FPKM值分别为:
接下来我们就可以计算FPKM的均值了。我们得到:
样本X在这5个基因上的FPKM均值FPKM_mean = 5,680;
样本Y在这5个基因上的FPKM均值FPKM_mean = 161,840
看到了吗?它们根本不同,而且差距相当大,那么究竟为什么会有如此之大的差异?难道是因为我故意构造出来的例子所造成的吗?当然不是,我们分析一下就会发现,这是完全是其数学公式所决定的。
首先,我们可以把FPKM的计算式拆分成如下两部分。
第一部分的统计量是对一个基因转录本数量的一个等价描述(虽然严格来讲也没那么等价):
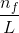
第二部分的统计量是测序获得的总有效Fregment数量的百万分之一:
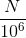
看到了吗?FPKM其实就只是这两部分的商!这有道理吗?分开来看它们似乎都有点道理,但是合起来的时候其实很没逻辑。
尤其是第二部分(N/10^6),本来式子的第一部分是为了描述一个基因的转录本数量,那么正常来讲,第二部分就应该是样本的转录本总数量(或至少是其总数量的等价描述)才能形成合理的比例关系,而且可以看出来FPKM/RPMK是有此意的,这本来就是这个统计量的目的。
可是,它却失败了!
N/10^6的大小其实是由RNA-seq测序深度所决定的,并且是一个和总转录本数量无直接线性关系的统计量——N与总转录本数量之间的关系还受转录本的长度分布所决定,而这个分布往往在不同样本中是有差异的!
比如,在有些基因中,虽然有效比对到基因的Fregment数是相等的,但一般来说长度越长的基因,其被转录的次数就越少。那也就是说,N必须将各个被转录的基因的长度考虑进去才能正确描述总体的转录本数!而FPKM/RPKM显然没有做到这一点,这便是FPKM和RPKM出错的内在原因。
那么应该是用什么样的统计量才合适?
其实,通过以上分析,我们已经可以确定一个更加合理的统计量来描述RNA转录本的丰度了。
这个统计量在2012年所发表的一篇讨论RPKM的文章(RPKM measure is inconsistent among samples. Wagner GP, Kin K, Lynch VJ. Theory Biosci. 2012.)中就被提出来了,称之为TPM —— Transcripts Per Million,它的计算是:
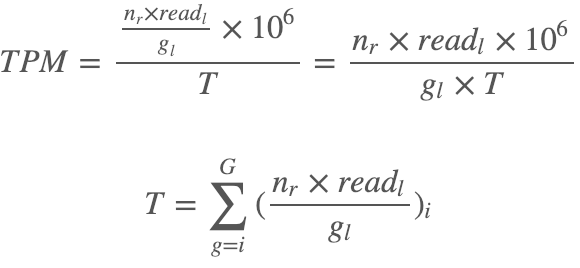
其中,read_l是比对至基因g的平均read长度,g_l 是基因g的外显子长度之和(这里无需将其除以1000,这是没必要的)。在不考虑read剪切的情况下,read_l这个值是测序长度,它往往都是一个固定值(如100bp或者150bp等),所以我们也可以将read_l统一消掉,这时分子就会蜕变成RPKM计算式的第一部分,只是留着会更好。
这样子,整个统计量就很好理解了,分子是某个基因g的转录本数(或者说是转录本数量的等价描述),分母为样本中总转录本的数量,两者的比值TPM——便是基因g的转录本丰度!并且,简单计算之后我们就可以发现TPM的均值是一个独立于样本之外的恒定值,它等于:
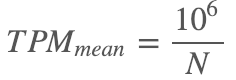
这个值刚好是r_mean的一百万倍,满足等价描述的关系。
我们仍然通过上面的例子来作进一步的说明,为简单起见我只把fregment换为read,其他数字都一样,并且统一假设read_l都是一样的:
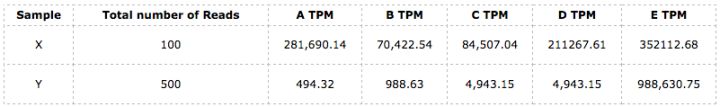
接着,我们可以分别计算样本X和Y的TPM_mean,并且很明显它们都是
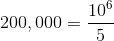
而且,经过这样的标准化之后,X和Y就处于同样的一个标准上了,此刻,在同一个坐标系下的差异比较才是真正有意义的。
既然FPKM/RPKM是错的,那为什么大家还在用,而且还真找到了(能被实验所验证)的有价值结果呢?
关于对于这个问题,我也思考过。而且我们都知道2008年那篇提出RPKM的文章也用实验结果来证明RPKM是一个合适的统计量,符合qPCR的验证结果。
但归根到底,眼见未必为实,实验也是表象的一个结果,我们应该回到其本质意义和原理上去思考。
FPKM/RPKM之所以看起来会是一个合适的值,我想主要原因有三:
其一,它们和TPM之间存在一定的正比关系,如下。通过比较它们各自的数学计算式就可以看出来(以RPKM的计算为例):
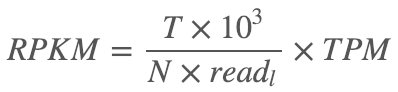
可以看到,在RPKM的公式中,原来TPM是其中的一个因子,而且在同一个样本内部由于T、N和read_l实际上都是定值——也就是说式子的第一个因子此时变成常数了!这就使得在同个样本内的RPKM和TPM是可以恒等转换的,可是这个情况在不同样本之间就不成立了!
因为不同样本之间的转换因子大小是不一样的!还是上面的例子,对于样本X,TPM转换到RPKM的转换因子为:0.0284,但样本Y的转换因子却是:0.8092。而由于这个标准的改变,会导致其原本所要描述的“转录本丰度”变得不可比较。
然,这其实还不是最根本的原因,更本质的原因是,这个转换会对本来已经正确标准化了的结果,再次做了一次无意义的不等变换,最终导致了结果不可解释。如何理解呢?后文会有补充,这里先简单说一下:这个数学转换式子仅是告诉了我们这样子来计算是可行的,但是在RNA-seq的实际应用场景中,它其实是无生物(或物理)意义的;
其二,实验验证的精度是有限的,常用的qPCR也只能给出定性的比较结果,而且实验验证也未必总能成功;
其三,习惯了,形成了路径依赖,大家都这样做,所以也这样跟随,这应该是最不可取的一个情况。
总结
现在回过头来总结一下。事实上,FPKM/RPKM最大的问题就在于其无意义性。我们所要表达出来的任何统计量,它的变化都应该能对应到物理或生物过程中的变化,如果做不到这一点,那么这个统计量往往都是无意义的,用它得到的结果就算看起来符合预期也只不过是数值上的巧合,本质上是不可解释的。FPKM/RPKM的分母(N/10^6)并不具有任何形式的生物意义,它所能表达出来的这个量,只能代表测序深度的变化,而无法作为对生物基因表达量的描述,即无法代表(等价代表)样本中转录本的总量。
一个统计量该如何计算,说到底都只是一个“术”的问题,而我们应该尽可能在接近其本质意义的地方去确定。
FPKM/RPKM和TPM存在一定的正比关系,因此我们在使用FPKM/RPKM时,有些时候确实也能获得可以被实验所验证的“好”结果,但其实它是一个橡皮筋,它的单位刻度是会随着样本的不同而改变的。到头来,样本之间的差异分析实际上也只是在不同的标准下进行的,这样的比较就算得到了所谓的“好”结果,那又有什么意义?根本就是个错误的东西。
想想就是由于这种统计量,我们一定已经获得了许多的假阳性结果,同时也肯定错过了许多本来真正有意义的差异,真是弯路走尽却不自知,而且过程中还浪费了多少的心情和时间?
这篇文章:A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Briefings in Bioinformatics.10.1093/bib/bbs046. 对7种主要的RNA-seq标准化方法(但不包含本文提到的TPM)做了一个详细的比较,它用实际结果进行比较(不同于本文所用的数学方式)也得出了RPKM这个统计量是应该被摒弃的结论,因为它所描述出来的结果是最不合理的,其实所有类似于RPKM(包括FPKM)的统计量在描述转录本丰度的时候都应该被摒弃。
时至今日,这个反思的经历依然可以不断给我带来有益的启发,其中最重要的一点就是绝不盲从、绝不轻信,如果我不能理解其原理和来龙去脉,那么我宁可不用,这也是为什么后来我总是更愿意自己去把方法实现的一个重要原因。想起一句话:
What I cannot create, I do not understand.
- Richard P.Feynman(理查德.菲利普斯.费曼)

费曼
愿与你共勉。

参考文献:
http://bib.oxfordjournals.org/content/early/2012/09/15/bib.bbs046.full
http://www.ncbi.nlm.nih.gov/pubmed/22872506
----/ END /----
Nature重磅综述 |关于RNA-seq,你想知道的都在这
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集