
iMeta视频教程 | 代谢物溯源/微生物组与代谢组整合分析软件MetOrigin
点击蓝字 关注我们
代谢物溯源分析软件MetOrigin
超详细的视频教程

自2022年2月份发布,MetOrigin已拥有国内外170+用户,3000+访问量!
全文回顾:
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析
应很多用户的需求和建议,浙江大学医学院附属儿童医院--倪艳博士研究团队持续更新和优化MetOrigin软件分析功能和数据库,并升级云服务计算系统,提供更高效的分析和更好的用户体验!
MetOrigin 的访问链接是:http://metorigin.met-bioinformatics.cn/
视频教程
Bilibili: https://www.bilibili.com/video/BV1dT411w7dw/
文字解说
我们在浏览器输入这个网址进入MetOrigin首页,然后在右上角点击“Login”按钮进入登录页面,如果没有注册账号,您可以点击右下角“register”按钮通过邮箱注册一个账号,或者使用tester账号进行登录,此处我们使用tester账号进行演示。
进入软件,我们首先提供了“Quick Search”功能,实现对单个代谢物快速搜索。输入内容允许是KEGG ID,HMDB ID或是代谢物全称,以KEGG ID--“C02656”为例,搜索结果为两个表格,第一张表“Metabolic reactions”展示了该代谢物所参与的所有微生物代谢反应,第二张表“Related microbes” 罗列了这些反应所有相关的微生物及具体的酶。
接下来我们介绍主要的两个分析流程,MetOrigin 根据代谢物和微生物数据集的可用性以及研究目的提供两种类型的数据分析模式。Simple MetOrigin Analysis (SMOA)模式和Deep MetOrigin Analysis (DMOA)模式
● Simple MetOrigin Analysis (SMOA) 模式
我们先来介绍SMOA模式。从左侧菜单我们可以看出,该模式包括5个步骤:
首先来到第一步,加载数据,我们需要选择正确的宿主信息,如人、小鼠、大鼠等,这里我们选择默认选项 -- human,然后在颜色设置框中,我们可以自定义设置不同来源代谢物的颜色。接下来是上传数据,点击“Browse”按钮选择您的文件进行上传,或者点击“Load Example Data”按钮加载软件自带示例数据进行测试。SMOA分析仅需要上传这一个表格, 该表格必须至少包含“HMDBID”、“KEGGID”或“Name”中的其中一列(当然,提供的信息越完整越好,如果缺失某列,软件会自行匹配缺失的信息,另外为了防止输入的HMDBID是旧版的ID,软件会将其转换为最新版的ID,所以如果结果中您输入的HMDBID有缺失的话,您可以确认一下该ID是否在HMDB数据库中真实存在)。最后,您可以提供Diff列来表示代谢物的统计显著性,1表示显著,0表示不显著。如果缺少“Diff”列,那么所有代谢物都将被视为差异代谢物,会进入后续的功能富集分析中。
上传数据完成,我们进行第二步的代谢物来源分析,单击“Perform Analysis”按钮开始分析。可以看到,结果生成了条形图、维恩图以及一张表格,两张图都是总结来自宿主、微生物、共代谢或者其他来源的代谢物的个数,图片填充颜色由上一个页面统一设置。下方表格展示了每一个代谢物的具体来源,Links列则是标明了判断为该来源是基于哪些数据库,点击ID即可跳转到对应数据库查看具体信息。
接下来我们进行代谢物的功能分析,单击“Perform Analysis”按钮开始数据分析。上一步我们已经区分了代谢物的来源,这一步根据不同来源的差异代谢物进行通路富集分析,这里主要是微生物来源、宿主来源以及共代谢来源。左侧韦恩图,描述了各来源代谢物分别参与代谢通路的交并集情况。右侧的条形图展示每条通路通过超几何分布计算得到的P-value,这里P-value以0.05为底进行了Log转换,所以Y轴大于1对应的通路被认为统计学显著。下面的表格,展示以上所有代谢通路的具体信息,没有富集的通路则不予显示。
下一步为桑基网络分析,左侧是一个交互式表格,这个表格提供了来自微生物或共代谢的代谢通路列表,也就是上面功能分析的结果。您可以通过勾选单元格或者点击单元格右下角拖拽复制来删除或添加特定的通路,系统默认只勾选最显著的一条通路,只有标有底色的单元格可选,深色单元格代表的代谢通路为具有统计学意义的通路。点击“Perform Analysis”按钮开始分析,这里需要一些时间等待结果。分析完成后,我们可以看到下面生成了两个Tab,分别是微生物来源和共代谢来源的桑基图。按照每条通路--每个反应我们各绘制了一张图,如图所示,最右列展示了参加该反应的底物、产物以及酶,颜色分别为红色、绿色以及紫色,前面几列则是可能参与该反应的所有微生物的具体层级信息,颜色为随机生成。您可以使用左上角的“Level”复选框选择查看哪些分类层级的微生物,并使用滑动条修改图形大小。网络图中的节点都可以自由拖动,您可以调到一个满意的位置之后使用左上角相机按钮将这些图片保存为 svg 格式并下载到本地计算机。
分析完以上内容,SMOA模式就结束了,我们可以进入最后一步进行结果下载。点击“Download Analysis Result”按钮即可下载完整结果。如果需要PDF格式的图片,可在“SVG Converter”模块,将之前下载的SVG图片上传进行转换。
● Deep MetOrigin Analysis (DMOA) 模式
接下来我们介绍DMOA模式,刷新浏览器,开始一个新项目。来到“Load Data”页面,将分析模式切换为DMOA模式,可以看到该模式包含7个步骤,比SMOA模式多出两个分析:相关性分析和网络总结分析。
回到数据加载页面,同样的我们先进行宿主的选择和颜色设置, 由于DMOA模式后续涉及到统计分析,因此这里多了对上下调或者正负相关性关系的颜色设置。“Test Method”模块这里我们进行一下统计方法的选择,软件总共提供了两种统计方法:T test和 U test,如果选择Auto的话,软件则会根据每个代谢物或者微生物数据是否满足正态分布以及它们的方差齐性来自动选择统计方法。后面的P-value Cut-off 为设定的差异代谢物的阈值,默认为0.05,您可以根据您的需要进行相应的调整。
接下来上传数据,我们需要上传3个表格,我们加载示例数据进行演示。第一个表--样本信息表,需要代谢物分析的样本 ID、微生物分析的样本 ID 和样本分组信息。这个表是可以编辑的,例如重命名样本或更改分组,可以在“Included”列进行勾选或取消勾选来增删样本,单击“Save”按钮即可保存修改内容。注意,样本名请尽量只由字母、数字、下划线组成,当分组类型选择“Categorical”时,“Grouping”列只提供0或1输入(0表示对照组,1表示疾病组,之后的fold change计算为疾病组比上对照组),反之,当分组类型选择“Continuous”时,“Grouping”只允许输入一列连续性数值(如BMI),与此同时,上面的统计方法选项也将对应变为Spearman和Person,后续将根据相关性的P-value来筛选差异代谢物或微生物。
第二个表格为代谢物表,这个表格需要至少“HMDBID”、“KEGGID”或“Name”中的其中一组,然后是每个样品的定量值。需要注意的是,样本编号必须和样本信息表中的样本编号一致,如有缺失会报错弹窗提示。表格上方,可进行数据预处理参数设置,您可以根据需要切换到合适的方法。
最后一个表是微生物数据表,这个表至少需要一列具有正确列名的分类注释信息,例如界、门、纲、目、科、属和种。同样,样本ID必须与样本信息表中微生物组分析的样本ID一致。
数据上传完成,我们进行代谢物来源分析和功能分析,这两步和SMOA模式内容一致,在此跳过。接下来进行相关性分析,我们先在这里进行参数设置,选择相关性分析方法,MetOrigin 提供了三种经典的相关分析方法,包括 Spearman、Pearson 和MIC分析。后面两个是显著性标记,P-value小于0.05标记一个星号,小于0.01标记两个星号。注意,第一个 P-value将作为后续分析中代谢物与微生物之间相关性的阈值。点击这两个色块我们可以自定义热图的颜色。点击“Perform Analysis”按钮开始分析,结果是差异代谢物与差异微生物每个层级的相关性热图,默认显示显著性前50的变量,缺失某个层级的热图表明该层级没有具有统计学差异的微生物。左侧“Parameter Setting”按钮可以进行字体等的调节,拖拽图片右下角三角形可以对图片进行尺寸调节。
下一步到桑基网络图,选择关注的通路,执行分析。我们可以看到, DMOA除了提供一个BIO-Sankey外还提供一个STA-Sankey网络图。BIO-Sankey基于MetOrigin数据库,代表生物学上的意义,将所有涉及该反应的微生物列举了出来, 您可以勾选“Show identified microbes only”选项来只显示自己上传数据中的微生物。STA-Sankey则是基于统计学意义,根据相关性分析的结果,将与参与该反应的代谢物显著相关的微生物挑选出来进行展示,这里的阈值即为相关性分析时第一个P-value Cut-off,名称后带星号表示该微生物在MetOrigin数据库中是可以查到的。对于每个代谢反应,条带的宽度与参与特定代谢反应的微生物数量成正比,条带的红色或绿色都表示该微生物或代谢物在此次研究中的上下调关系,或它们之间的正负相关关系。颜色的深浅则表示是否显著上下调或者显著相关,具体可参照右上角图例。
最后一个分析,网络总结,左上角交互式表格操作同桑基网络图,不同的是,只有具有统计学意义的通路才会显示在该表格中(深色单元格可选)。选择通路,执行分析。获得微生物和代谢物相互作用的网络图。相应地总结了宿主、微生物和共代谢三种代谢网络的所有选中的反应及相关微生物。此处由于宿主没有富集到的通路,所以不予显示。可根据网络图左上角“Level”选项卡来切换关联的微生物层级,也可以根据“Microbe Pvalue Cutoff”来更改微生物筛选的阈值。网络图具体颜色含义详见软件右上角图。(注意:如果分析完成未出图,而前面相关性分析确有结果的,可能是因为微生物名称与MetOrigin库不一致)。
到此所有分析完成,同样的,您可以到下载页面下载完整结果。
联系方式:
如有任何问题或建议,请联系我们的技术支持(bioinformatics_group@aliyun.com),或本项目负责人倪艳博士(yanni617@zju.edu.cn)
Nilab课题组网页连接
:http://nilab.met-bioinformatics.cn/
更多推荐
(▼ 点击跳转)
iMeta封面 | 宏蛋白质组学分析一站式工具集iMetaLab Suite(加拿大渥太华大学Figeys组)
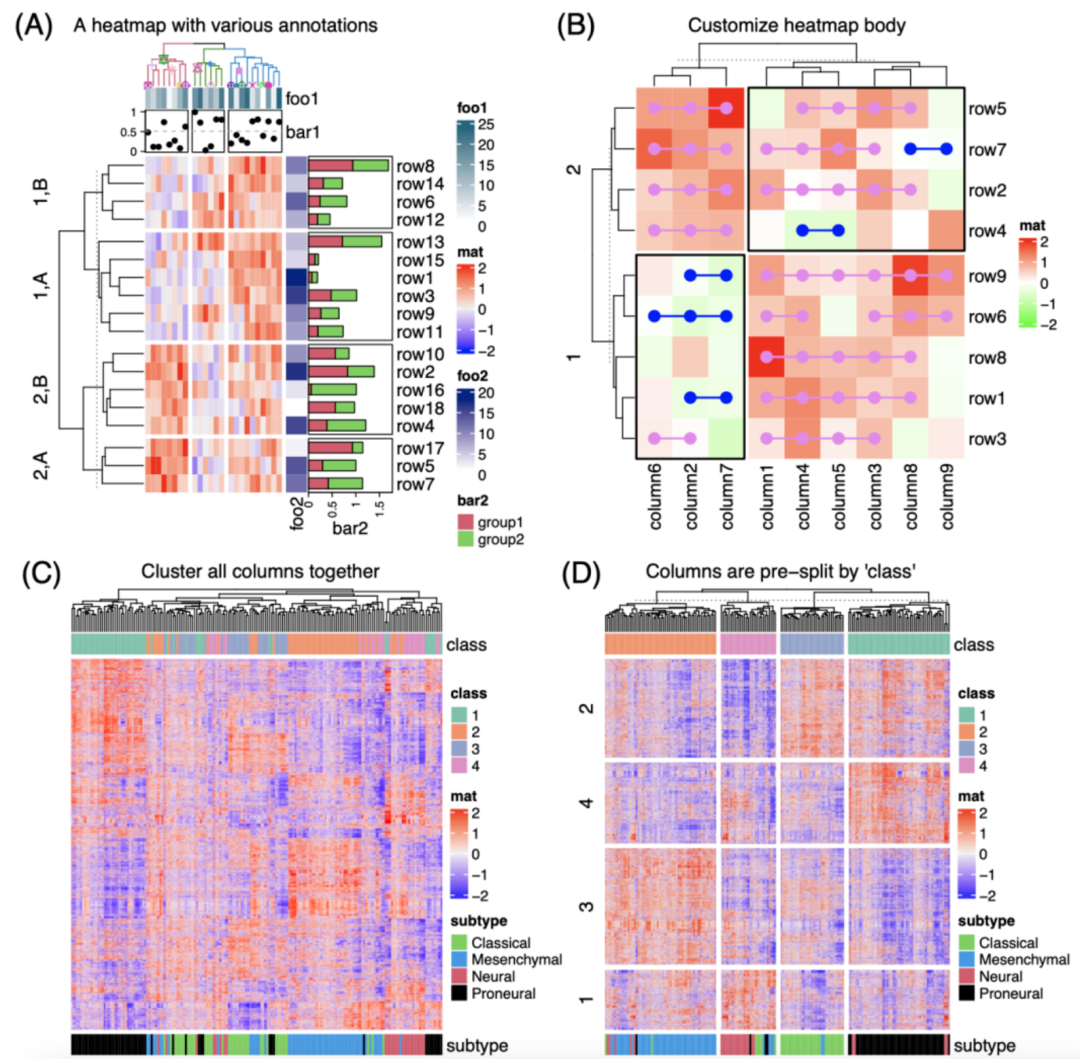
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法
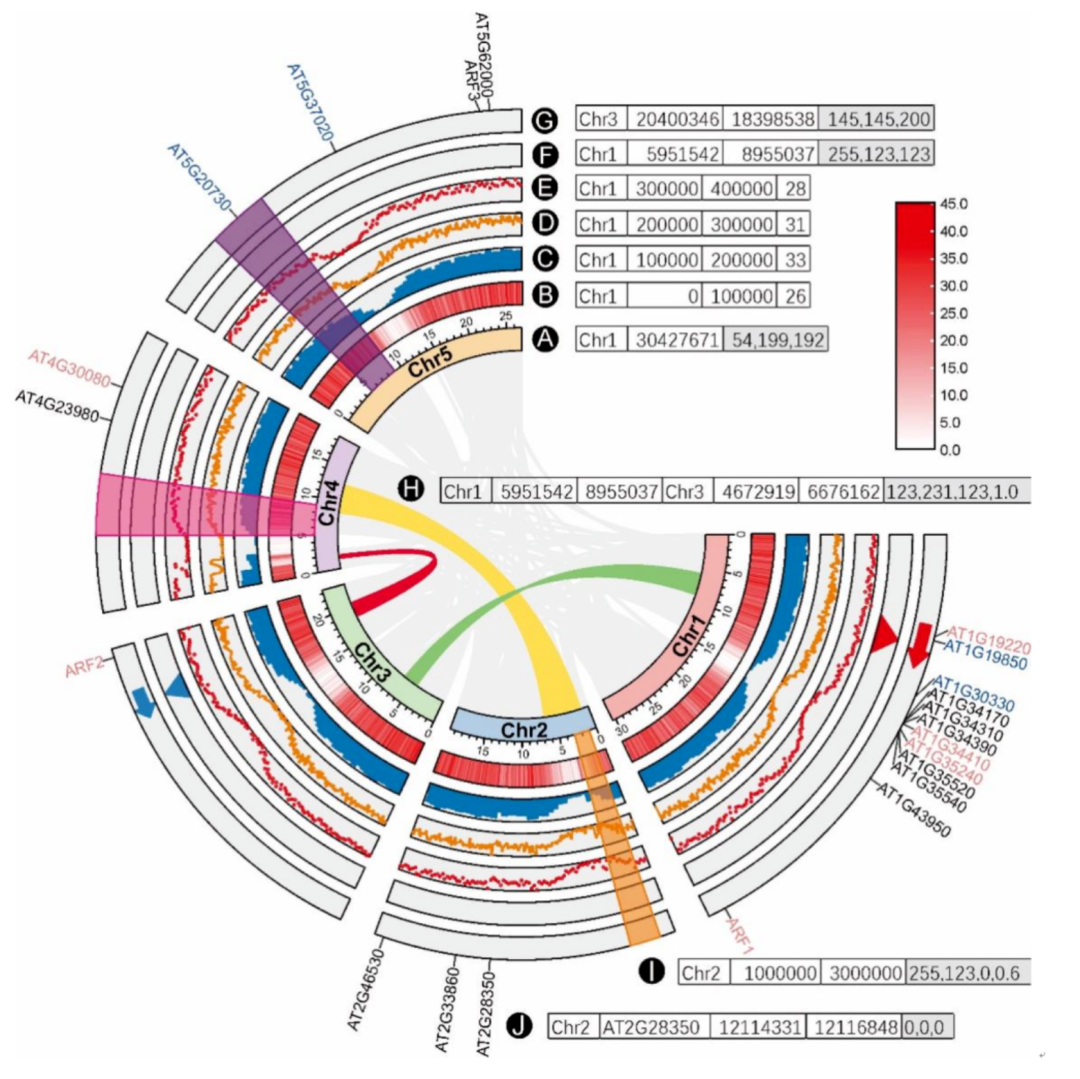
iMeta | 华南农大陈程杰/夏瑞等发布TBtools构造Circos图的简单方法
往期回顾

🔗:https://onlinelibrary.wiley.com/toc/2770596x/2022/1/1

🔗:https://onlinelibrary.wiley.com/toc/2770596x/2022/1/2
期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science
微信公众号
iMeta
责任编辑
微微