
IPython最初只有259行代码!Nature盘点了十项改变科学的代码

新智元报道
【新智元导读】从天文学到生态学,现代每一个伟大的科学发现背后都有一台计算机。对你来说,哪个计算机项目对你的影响最大?
Michael Levitt 是加利福尼亚州斯坦福大学的计算生物学家,他因在化学结构建模的计算策略方面的工作获得了2013年诺贝尔化学奖。他指出,今天的笔记本电脑的内存和时钟速度是他在1967年开始获奖工作时实验室制造的计算机的1万倍。
的确,计算机的发明极大的提升了效率,释放了生产力,也成为现代人不可不会的生存技能。
近日,Nature就整理了10项改变科学的计算机项目,arXiv、IPython等均在列。
下面是这10个项目的详细介绍:
语言先驱:Fortran编译器(1957年)
在1950年代这个计算机并不发达的时代,编程还是通过手工将电线连接成电路组完成的。当后来有了机器语言和汇编语言,用户可以用代码进行编程了,但即便省去了手上的工作,人们却面临另一个难题——需要对计算机结构有深入的了解,这让许多计算机专家也束手无策。
这种情况在20世纪50年代随着符号语言的发展而改变,特别是由John Backus和他的团队在加州圣何塞的IBM开发的「公式翻译」语言Fortran。
使用Fortran,用户可以使用人类可读的指令对计算机进行编程,例如x=3+5。然后,编译器会将这种指令转化为快速、高效的机器代码。

图:CDC 3600计算机,于1963年交付给科罗拉多州博尔德市的国家大气研究中心,在Fortran编译器的帮助下进行了编程
在早期,程序员使用打孔卡来输入代码,一个复杂的模拟可能需要数万个打孔卡。不过,新泽西州普林斯顿大学的气候学家Syukuro Manabe说,Fortran让非计算机科学家的研究人员也能使使用编程。「第一次,我们能够自己给的计算机编程,」Manabe说。他和他的同事们使用该语言开发了首批成功的气候模型之一。
如今Fortran已经发展到第八个十年了,它仍然被广泛地应用于气候建模、流体动力学、计算化学等,这些学科涉及复杂的线性代数,并需要强大的计算机来快速处理数字。由此产生的代码速度很快,而且仍然有很多程序员知道如何编写它。复古的Fortran代码库仍然活跃在实验室和全球的超级计算机上。
信号处理器:快速傅立叶变换(1965年)
当射电天文学家扫描天空时,他们捕捉到了随着时间变化的复杂信号的嘈杂声。为了了解这些无线电波的性质,他们需要看到这些信号作为频率的函数是什么样子。一种被称为傅里叶变换的数学可以帮到他们。可问题是,它的效率太低了,对于一个大小为N的数据集来说,就需要进行N2次计算。
1965年,美国数学家James Cooley和John Tukey研究出了一种加速方法:快速傅里叶变换(FFT),它利用递归(一种「分而治之」的编程方法,在这种方法中,一个算法反复地重新应用自己,将计算傅里叶变换的问题简化到只有N个对数2(N)步。随着N的增长,速度也会提高。
其实,在1805年,德国数学家高斯就研究出了它,但他从未发表过。而Cooley和Tukey的「再发现」,却开启了数字信号处理、图像分析、结构生物学等方面的应用。FFT已经实现了很多次,目前有一种流行的方案叫做FFTW。

图:Murchison Widefield Array的一部分夜景,这是西澳大利亚州的射电望远镜,使用快速傅里叶变换进行数据收集
分子目录员:生物数据库(1965年)
在过去的几十年里,数据库得到了大规模的发展,而且影响了很多领域,但是,最令人瞩目的还属生物学。
今天,我们看到庞大的基因组和蛋白质数据库,这起源于Margaret Dayhoff的工作,她是美国物理化学家,也是生物信息学领域的先驱。
在20世纪60年代初,当生物学家们努力拆解蛋白质的氨基酸序列时,Dayhoff整理了这些信息,以找寻不同物种之间进化关系的线索。
她与另外三位合著者的《蛋白质序列和结构图谱》(Atlas of Protein Sequence and Structure)于1965年首次出版,这本书描述了当时已知的65种蛋白质的序列、结构和相似性。这本图册中的数据,演化为后来的蛋白质信息资源数据库 PIR( Protein Information Resource ) 。
其他「计算机化」的生物数据库也随之而来。蛋白质数据库于1971年上线,今天它详细记录了17万多个大分子结构。1981年,加州大学圣地亚哥分校的进化生物学家Russell Doolittle创建了另一个名为Newat的蛋白质数据库。而1982年,该数据库发布,成为美国国立卫生研究院维护的DNA档案库GenBank。
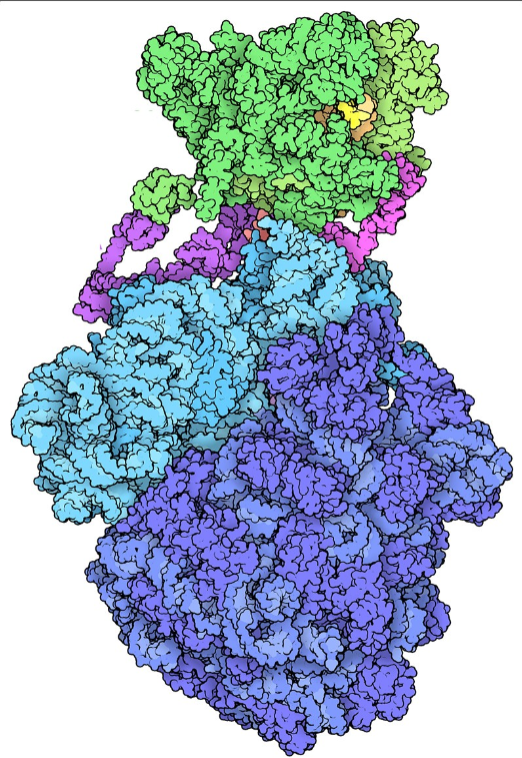
图:蛋白质数据库有一个超过17万个分子结构的档案,包括这个细菌的 "表达组",它结合了RNA和蛋白质合成的过程
1983年7月,这样的资源在证明了其存在的价值,当时,由伦敦帝国癌症研究基金的蛋白质生物化学家迈克尔-沃特菲尔德和杜利特尔领导的独立团队,报告了一种特殊的人类生长因子序列与一种猴子身上致癌的病毒蛋白质之间的相似性。这一观察结果提出了一种由病毒引起肿瘤发生的机制--通过模仿一种生长因子,病毒会诱导细胞不受控制地生长。
除了设计实验来测试特定的假设,研究人员还可以挖掘公共数据集,寻找那些实际收集数据的人可能从未想到的联系。
当不同的数据集被链接在一起时,这种力量就会急剧增长--1991年,NCBI的程序员通过Entrez实现了这一目标,Entrez是一个允许研究人员从DNA到蛋白质再到文献,然后再返回的工具。
位于马里兰州贝塞斯达的NCBI现任代理主任Stephen Sherry在研究生时期就使用过Entrez。「我记得当时我觉得它很神奇。」他说。
预测领先者:大气循环模型(1969年)
第二次世界大战结束时,计算机先驱约翰-冯-诺依曼开始将几年前还在计算弹道轨迹和武器设计的计算机转向天气预报问题。Manabe解释说,在此之前,"天气预报只是经验性的",利用经验和直觉来预测接下来会发生什么。相比之下,冯-诺依曼的团队 "试图根据物理定律进行数值天气预测"。
位于新泽西州普林斯顿的美国国家海洋和大气管理局地球物理流体动力学实验室的建模系统部负责人Venkatramani Balaji说,几十年来,这些方程已经为人所知。但早期的气象学家无法实际解决这些问题。
要做到这一点,需要输入当前的条件,计算它们在短时间内会如何变化,然后重复。这个过程非常耗时,以至于在天气本身到来之前,数学都无法完成。1922年,数学家刘易斯-弗莱-理查德森花了几个月的时间来计算德国慕尼黑的6小时预报。根据一份历史资料,结果是 "非常不准确",包括 "在任何已知的陆地条件下都不可能发生 "的预测。
计算机使这个问题变得容易解决。
20世纪40年代末,冯-诺依曼在普林斯顿高等研究院成立了他的天气预测小组。1955年,第二个小组:地球物理流体动力学实验室,开始了他所谓的 "无限预报"--即气候建模的工作。
Manabe于1958年加入气候建模团队,开始研究大气模型;他的同事Kirk Bryan则研究海洋模型。1969年,他们成功地将两者结合起来,创造了2006年《自然》杂志所说的科学计算的 "里程碑"。
今天的模型可以将地球表面划分为25×25公里的方块,将大气层划分为几十层。相比之下,Manabe和Bryan的海洋-大气组合模型5使用了500公里的方块和9个层次,只覆盖了地球的六分之一。不过,Balaji说,"那个模型还是做得很好",使该团队首次能够在硅片中测试二氧化碳水平上升的影响。
数学运算器:BLAS(1979年)
科学计算通常涉及使用向量和矩阵进行相对简单的数学运算。它们的数量实在是太多了。但在20世纪70年代,还没有一套普遍认同的计算工具来执行这些运算。因此,在科学领域工作的程序员会把时间花在设计高效的代码来进行基础数学运算上,而不是专注于科学问题。
编程世界需要的是一个标准。在1979年,它得到了一个标准:基本线性代数子程序,或BLAS6。
这个标准一直发展到1990年,为向量数学和后来的矩阵数学定义了几十个基本例程。位于诺克斯维尔的田纳西大学的计算机科学家Jack Dongarra说,实际上,BLAS将矩阵和矢量数学还原成了和加减法一样基本的计算单位。
 图:Cray-1超级计算机:在1979年推出BLAS编程工具之前,没有线性代数标准供研究人员在加州劳伦斯利弗莫尔国家实验室的Cray-1超级计算机等机器上工作。
图:Cray-1超级计算机:在1979年推出BLAS编程工具之前,没有线性代数标准供研究人员在加州劳伦斯利弗莫尔国家实验室的Cray-1超级计算机等机器上工作。
德克萨斯大学奥斯汀分校的计算机科学家Robert van de Geijn说,BLAS "可能是为科学计算定义的最有影响的接口"。除了为常见功能提供标准化的名称外,研究人员还可以确保基于BLAS的代码在任何计算机上以同样的方式工作。该标准还使计算机制造商能够优化BLAS的实现,使其在硬件上快速运行。
40多年过去了,BLAS代表了科学计算堆栈的核心,是科学软件的代码。华盛顿特区乔治华盛顿大学的机械和航空航天工程师Lorena Barba称其为 "五层代码中的机械"。
显微镜必备: NIH Image(1987)
20世纪80年代早期,程序员 Wayne Rasband 在位于马里兰州贝塞斯达的美国国立卫生研究院研究所的脑成像实验室工作。研究小组使用扫描仪对 x 射线胶片进行数字化处理,但无法在电脑上进行显示或分析。所以拉斯班德写了一个程序来做这件事。
这个程序最初是专门为一台价值15万美元的 PDP-11小型计算机设计的,这是一台安装在机架上的非个人电脑。然后,在1987年,苹果公司发布了 Macintosh II,这是一个更友好、更实惠的选择。拉斯班德说: “在我看来,很明显,作为一种实验室图像分析系统,这种方法会工作得更好。”他将自己的软件移植到新平台上,并重新命名,建立了一个图像分析生态系统。
NIH Image及其后代使研究人员能够在任何计算机上查看和量化几乎任何图像。
这个软件系列包括 ImageJ,这是一个由 Rasband 为 Windows 和 Linux 用户编写的基于 java 的版本,以及 Fiji,一个由 Pavel tomanchak 的团队在德国德累斯顿的 Max Planck 分子细胞生物学和遗传学研究所开发的 ImageJ 分发版,其中包括关键的插件。“ ImageJ 无疑是我们拥有的最基础的工具,”计算生物学家贝丝 · 西米尼说,“我从来没有和使用过显微镜但没有用过 ImageJ 或 Fiji的生物学家交谈过。”

图:mageJ 工具在一个插件的帮助下,可以在显微镜图像中自动识别细胞核
序列搜索器: BLAST (1990)
也许没有什么比把软件名称变成一个动词更能说明文化相关性了。一提到搜索,就会想到谷歌。而对于遗传学,则会想到 BLAST。
进化的变化被蚀刻在分子序列中,如替换、删除、缺失和重排。通过寻找序列之间的相似性,尤其是蛋白质之间的相似性,研究人员可以发现进化关系,并深入了解基因功能,其中的关键就在于迅速和全面地构建内容快速膨胀的分子信息数据库。
1978年,戴霍夫为解决这个谜题提供了关键的进展。她设计了一个点接受突变矩阵(point accepted mutation matrix),研究人员可以根据两种蛋白质序列的相似程度,以及它们之间的进化距离来评估它们之间的关系。
1985年,夏洛茨维尔弗吉尼亚大学的 William Pearson 和 NCBI 的 David Lipman 引入了 FASTP 算法,该算法结合了 Dayhoff 的矩阵和快速搜索的能力。
多年以后,Lipman 与 NCBI 的 Warren Gish 和史蒂文·阿尔茨契尔,宾夕法尼亚州立大学的 Webb Miller 和图森亚利桑那大学的 Gene Myers 一起开发出了一种更加强大的改进技术: BLAST (Basic Local Alignment Search Tool)。BLAST 于1990年发布,将处理快速增长的数据库所需的搜索速度与提取进化上更为遥远的匹配结合起来。同时,这个工具可以计算出这些匹配发生的概率有多大。
结果是令人难以置信的快速,阿特舒尔说。“你可以进行搜索,喝一口咖啡,搜索就完成了。”但更重要的是,它很容易使用。在数据库通过邮寄更新的时代,Gish 建立了一个电子邮件系统,后来又建立了一个基于网络的架构,允许用户在 NCBI 的计算机上远程进行搜索,从而确保他们的搜索结果始终是最新的。
这个系统为当时刚刚起步的基因组生物学领域提供了一个变革性的工具——一种根据未知基因的相关基因推算出未知基因可能出现的可能性。
预印本平台:arXiv.org(1991)
20世纪80年代末,高能物理学家经常将他们提交的手稿的副本邮寄给同事,请其发表评论,但只发给少数人。物理学家保罗 · 金斯帕格在2011年写道: “那些处于食物链底层的人依赖于那些一线研究人员的成果,而那些有抱负的非精英研究机构的研究人员常常完全置身于特权圈之外”。
1991年,当时在新墨西哥州洛斯阿拉莫斯国家实验室的 Ginsparg 写了一封电子邮件自动应答,以平衡竞争环境。
订阅者每天收到预印本的列表,每个列表与一个文章标识符相关联。通过一封电子邮件,世界各地的用户可以从实验室的计算机系统提交或检索一篇文章,获得新文章的列表,或者按作者或标题进行搜索。
起初,金斯帕格的计划是将文章保留三个月,并将内容限制在高能物理学界。但一位同事说服他无限期保留这些文章。从此,论文如潮水般涌来。
到了1993年,Ginsparg 把这个系统移植到了互联网上,1998年,他把它命名为 arxiv. org。
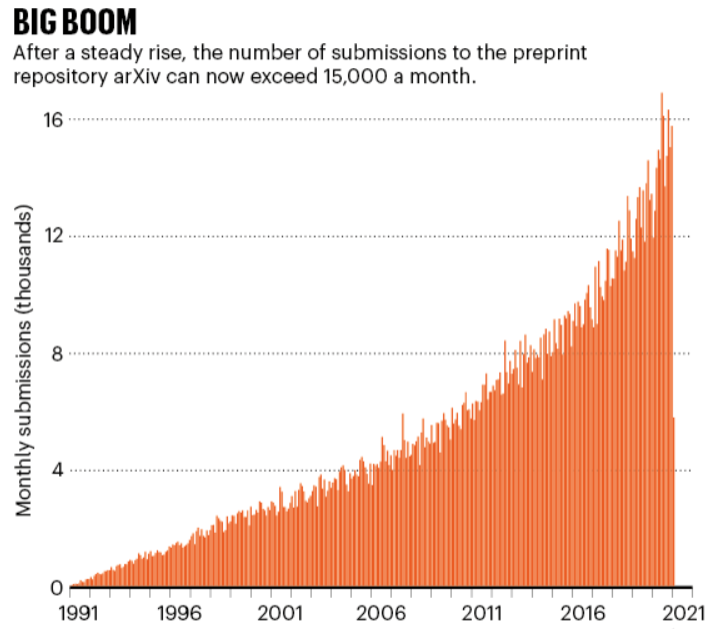
现在,arXiv 已经成立30年了,它收藏了大约180万份预印本,而且都是免费提供的,每个月下载量达到3000万次,提交量超过15000份。
这个系统为研究人员提供了一种快速方便的方式,可以通过一个标识,显示他们在什么时候做了什么,避免了在传统期刊上进行同行评审所需的麻烦和时间。
数据浏览器: IPython Notebook (2011)
2001年,Fernando Pérez 还是一名“寻找拖延症”的研究生,当时他决定采用 Python 的一个核心组件。
Python是一个可解释性语言,这意味着程序是逐行执行的。程序员可以使用一种称为读取-评估-打印循环(REPL:read–evaluate–print loop )的计算调用和响应工具,在这种工具中,他们输入代码,然后由解释器执行代码。
REPL 允许快速的探索和迭代,但是 pérez 指出 Python 不是为科学而构建的。例如,它不允许用户轻松地预加载代码模块,也不允许打开数据可视化。于是佩雷斯写下了他自己的版本。
其结果就是 IPython,这是一个“交互式”Python 解释器,佩雷斯在2001年12月公布了它的全部259行代码。
十年后,佩雷兹与物理学家布莱恩 · 格兰杰和数学家埃文 · 帕特森一起,将这个工具移植到了网络浏览器上,发布了 IPython Notebook,并开启了一场数据科学革命。
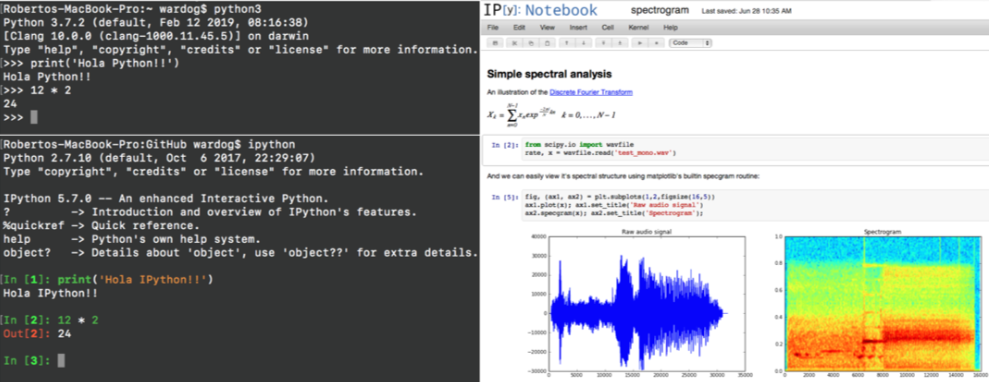
与其他计算型notebook一样,IPython Notebook 将代码、结果、图形和文本合并到一个文档中。但与其他此类项目不同的是,IPython Notebook 是开源的,邀请了大量开发者社区的贡献。它还支持 Python这种科学家们喜欢的语言。2014年,IPython 演变成了“Jupyter”,支持100多种语言,允许用户在远程超级计算机上探索数据,就像在自己的笔记本电脑上一样轻松。
《自然》杂志在2018年写道: “对于数据科学家来说,Jupyter 已经成为了行业标准”。当时,GitHub 代码共享平台上有250万 Jupyter notebooks; 如今,已有近1000万,其中包括记录2016年引力波发现和2019年黑洞成像的notebooks。佩雷斯表示: “我们为这些项目做出了一点贡献,这是非常值得的。”
快速学习器: AlexNet (2012)
人工智能通常有两种类型。一种使用编码规则,另一种使计算机通过模拟大脑的神经结构来“学习”。
计算机科学家Geoffrey Hinton曾表示,几十年来,人工智能研究人员一直认为后一种方法是“无稽之谈”。2012年,Hinton 的研究生 Alex Krizhevsky 和 Ilya sutskiver 证明了事实并非如此。
在 ImageNet比赛中,研究人员在一个包含100万张日常物品图像的数据库中训练人工智能,然后在一个单独的图像集上测试得出的算法。Hinton说,当时,最好的算法错误分类了大约四分之一。Krizhevsky 和 sutskiver 的 AlexNet 是一种基于神经网络的“深度学习”算法,它将错误率降低到了16% 。
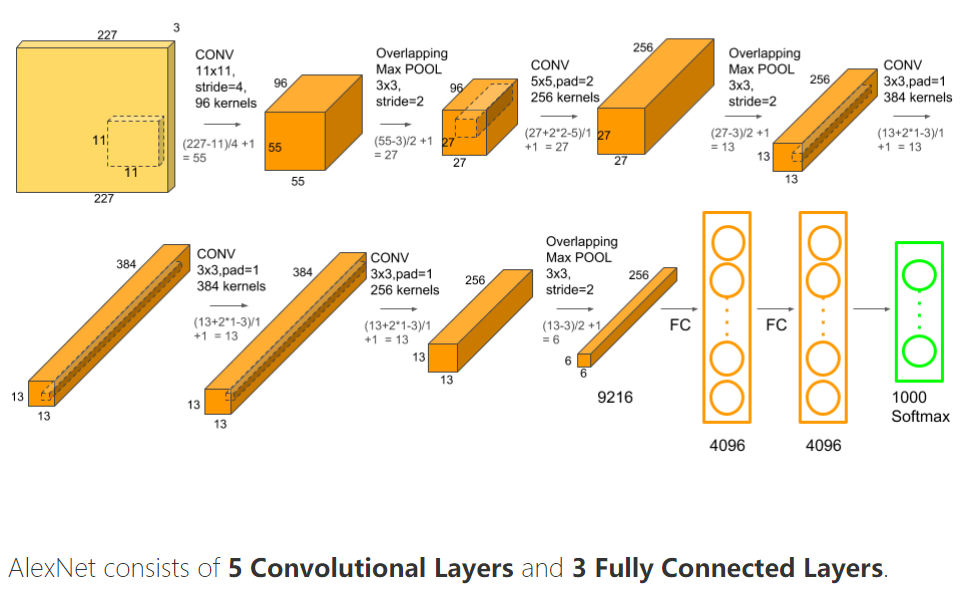
Hinton表示,该团队在2012年的成功反映了大量训练数据集、出色的编程和新出现的图形处理单元(最初设计这些处理器是为了加速计算机视频性能)的强大能力的结合。“突然之间,我们可以将(算法)运行速度提高30倍,或者在30倍于此的数据上学习。”
AlexNet的胜利预示着深度学习在实验室、医学等领域的兴起。这就是为什么手机能够理解语音查询,也是为什么图像分析工具能够轻而易举地在显微照片中挑出细胞。这就是为什么 AlexNet 在许多工具中占有一席之地,而这些工具从根本上改变了科学,以及世界。
参考链接:
https://www.nature.com/articles/d41586-021-00075-2
