R包ggseqlogo |绘制序列分析图
前言
NGS系列文章包括NGS基础、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述))、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step) - Limma差异分析、火山图、功能富集)等内容。
简介
在生物信息分析中,经常会做序列分析图(sequence logo),这里的序列指的是核苷酸(DNA/RNA链中)或氨基酸(在蛋白质序列中)。sequence logo图是用来可视化一段序列某个位点的保守性,据根提供的序列组展示位点信息。常用于描述序列特征,如DNA中的蛋白质结合位点或蛋白质中的功能单元。
实现以上可视化过程的工具有很多,本文介绍一个使用起来非常简单,不拖泥带水的R包ggseqlogo,只要你根据此包要求的数据格式上传一堆DNA序列或者氨基酸序列,再根据现成的命令流程就能画出logo图。
安装到作图的代码如下:
安装
安装方式有两种
#直接从CRAN中安装
install.packages("ggseqlogo")
#从GitHub中安装
devtools::install.github("omarwagih/ggseqlogo")数据加载
ggseqlogo提供了测试数据ggseqlogo_sample。
#加载包
library(ggplot2)
library(ggseqlogo)
#加载数据
data(ggseqlogo_sample)ggseqlogo_sample数据集是一个列表,里面包含了三个数据集:
seqs_dna:12种转录因子的结合位点序列
pfms_dna:四种转录因子的位置频率矩阵
seqs_aa:一组激动酶底物磷酸化位点序列
#seqs_dna
head(seqs_dna)[1]## $MA0001.1
## [1] "CCATATATAG" "CCATATATAG" "CCATAAATAG" "CCATAAATAG" "CCATAAATAG"
## [6] "CCATAAATAG" "CCATAAATAG" "CCATATATGG" "CCATATATGG" "CCAAATATAG"#pfms_dna
head(pfms_dna)[1]## $MA0018.2
## [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8]
## A 0 0 11 0 1 0 2 8
## C 1 1 0 9 0 3 7 0
## G 1 10 0 2 10 0 1 1
## T 9 0 0 0 0 8 1 2#seqs_aa
head(seqs_aa)[1]## $AKT1
## [1] "VVGARRSSWRVVSSI" "GPRSRSRSRDRRRKE" "LLCLRRSSLKAYGNG"
## [4] "TERPRPNTFIIRCLQ" "LSRERVFSEDRARFY" "PSTSRRFSPPSSSLQ"外部数据读入
也可以用自己的数据集,支持两种格式,序列和矩阵。
# 长度为7的motif。每一行为一条序列,长度相同,每一列的碱基组成代表对应位置的碱基偏好性。
fasta = "ACGTATG
ATGTATG
ACGTATG
ACATATG
ACGTACG"
fasta_input <- read.table(fasta, header=F, row.names=NULL)
fasta_input <- as.vector(fasta_input$V1)
# 长度为5的motif矩阵示例,每一列代表一个位置,及碱基在该位置的出现次数。共4行,每一行代表一种碱基
matrix <- "Base 1 2 3 4 5
A 10 2 0 8 1
C 1 12 1 2 3
G 4 0 9 1 1
T 0 0 0 1 9
"
matrix_input <- read.table(matrix, header=T, row.names=1)
matrix_input <- as.matrix(matrix_input)可视化
ggplot()+geom_logo(seqs_dna$MA0001.1)+theme_logo()
ggseqlogo提供了一个直接绘图的函数ggseqlogo(),这是一个包装函数。下面命令结果同上面的。
ggseqlogo(seqs_dna$MA0001.1)输入格式
ggseqlogo支持以下几种类型数据输入:
序列
矩阵
下面是使用数据中的位置频率矩阵生成的seqlogo
ggseqlogo(pfms_dna$MA0018.2)
方法
ggseqlogo通过method选项支持两种序列标志生成方法:bits和probability。
p1 <- ggseqlogo(seqs_dna$MA0001.1, method="bits")
p2 <- ggseqlogo(seqs_dna$MA0001.1, method="prob")
gridExtra::grid.arrange(p1,p2)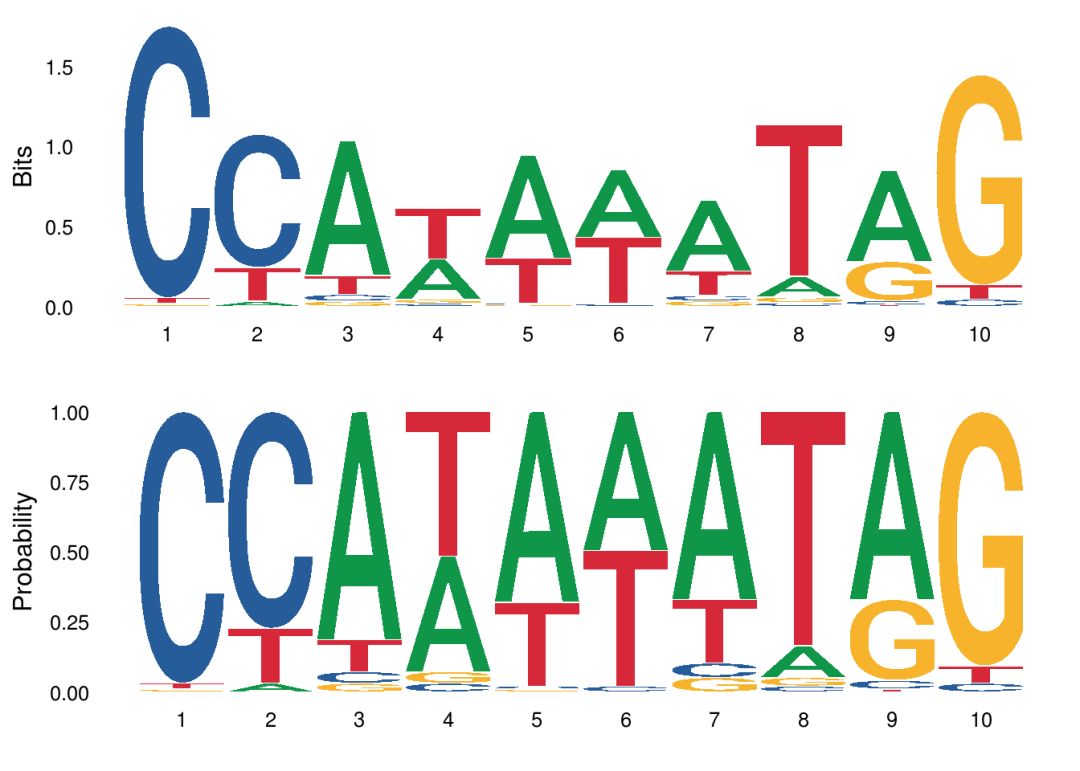
序列类型
ggseqlogo支持氨基酸、DNA和RNA序列类型,默认情况下ggseqlogo会自动识别数据提供的序列类型,也可以通过seq_type选项直接指定序列类型。
ggseqlogo(seqs_aa$AKT1, seq_type="aa")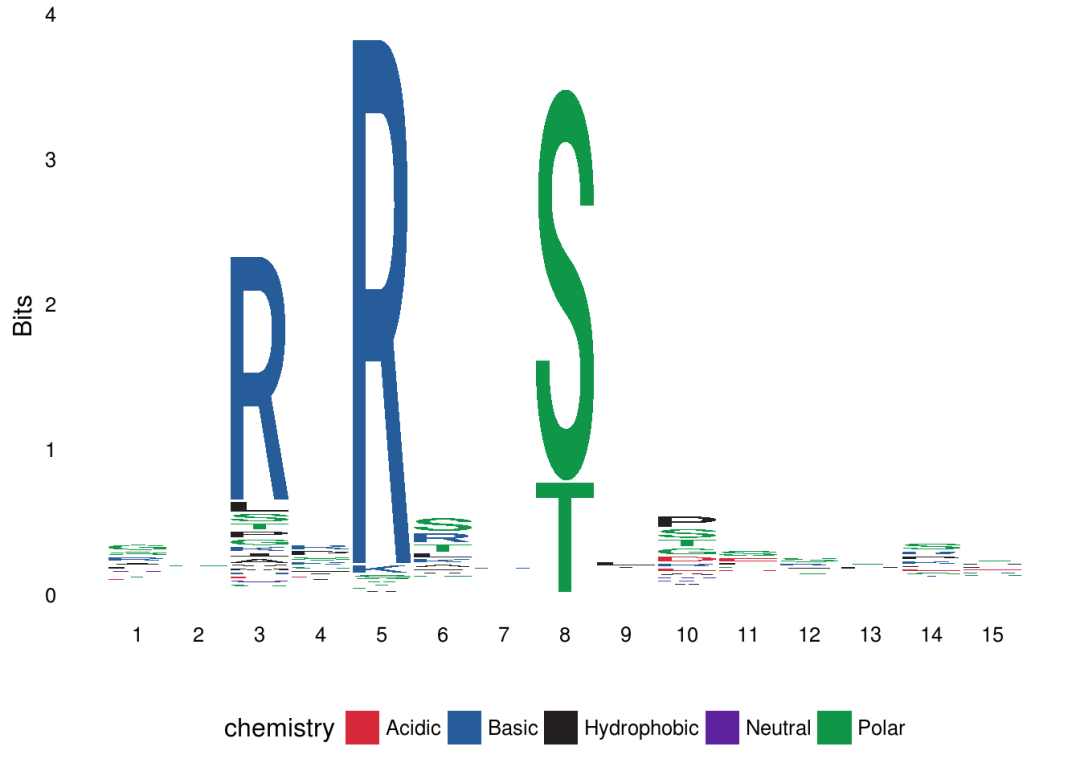
自定义字母
通过namespace选项来定义自己想要的字母类型
#用数字来代替碱基
seqs_numeric <- chartr("ATGC", "1234", seqs_dna$MA0001.1)
ggseqlogo(seqs_numeric, method="prob", namespace=1:4)
配色
ggseqlogo可以使用col_scheme参数来设置配色方案,具体可参考?list_col_schemes
ggseqlogo(seqs_dna$MA0001.1, col_scheme="base_pairing")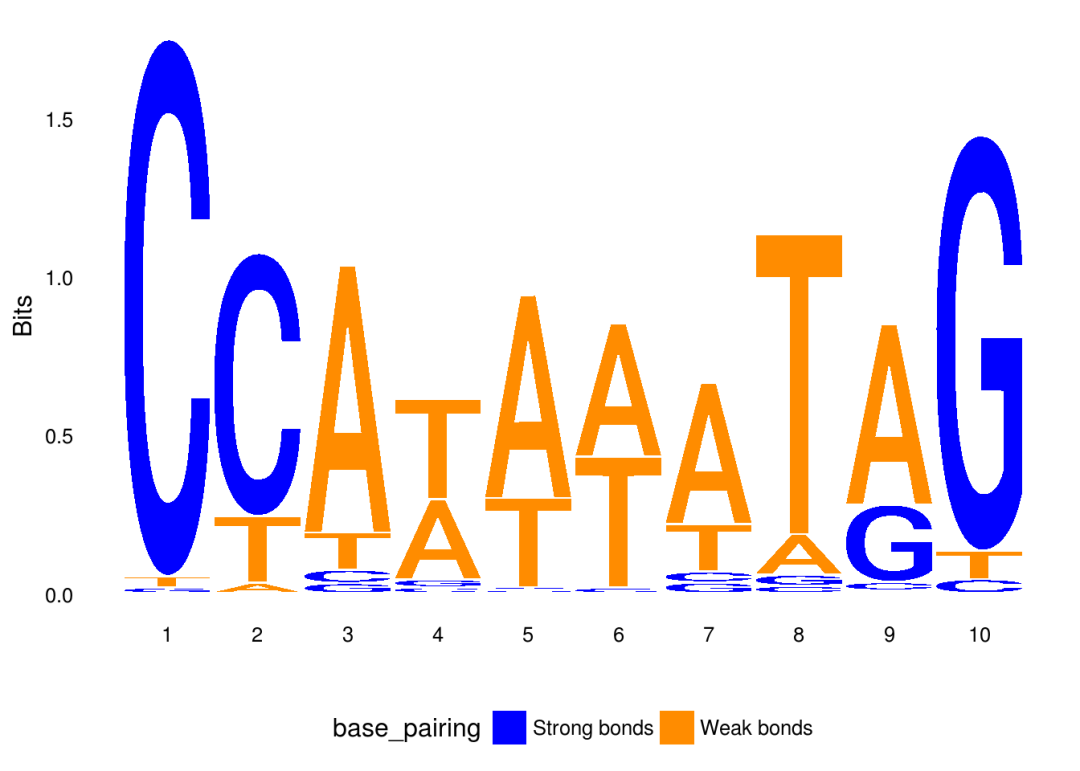
自定义配色
ggseqlogo提供函数make_col_scheme来自定义离散或者连续配色方案
离散配色
csl <- make_col_scheme(chars = c("A","T", "C", "G"), groups = c("gr1","gr1", "gr2","gr2"), cols = c("purple","purple","blue","blue"))
ggseqlogo(seqs_dna$MA0001.1,col_scheme=csl)
连续配色
cs2 <- make_col_scheme(chars = c("A", "T", "C", "G"), values = 1:4)
ggseqlogo(seqs_dna$MA0001.1, col_scheme=cs2)
同时绘制多个序列标志
ggseqlogo(seqs_dna, ncol = 4)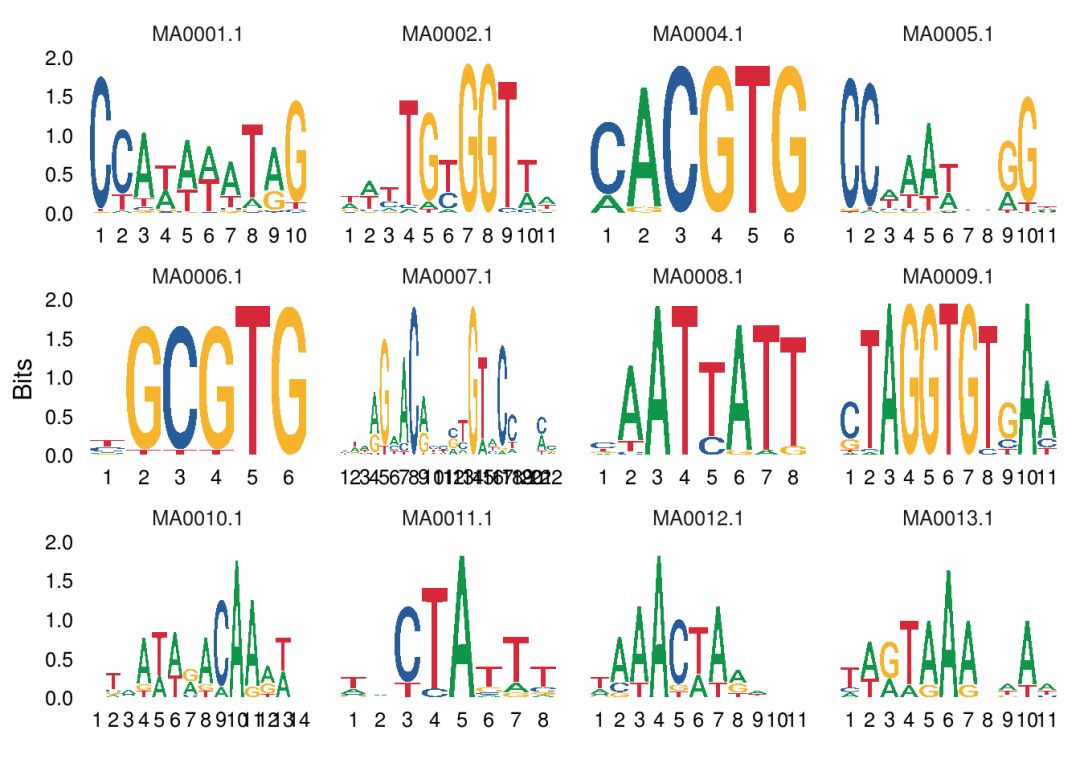
上述命令实际上等同于
ggplot()+geom_logo(seqs_dna)+theme_logo()+
facet_wrap(~seq_group,ncol = 4,scales = "free_x")自定义高度
通过创建矩阵可以生成每个标志的高度,还可以有负值高度
set.seed(1234)
custom_mat <- matrix(rnorm(20), nrow = 4, dimnames = list(c("A","T","C", "G")))
ggseqlogo(custom_mat,method="custom",seq_type="dna")+
ylab("my custom height")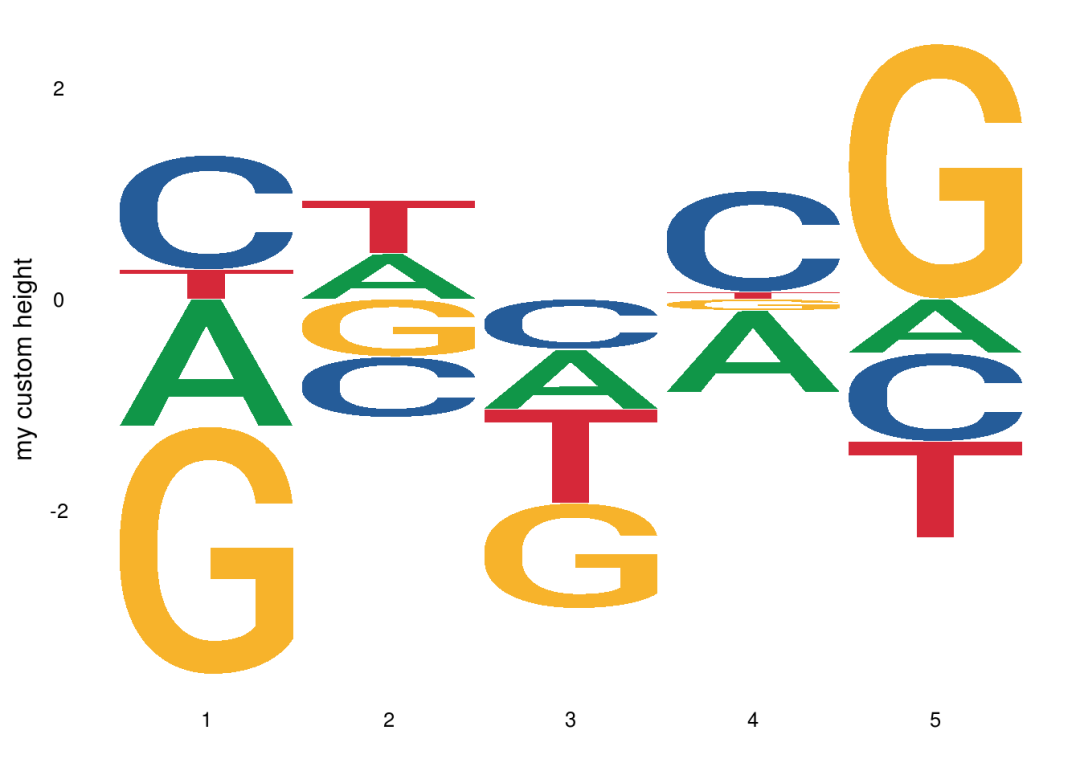
字体
可以通过font参数来设置字体,具体可参考?list_fonts
fonts <- list_fonts(F)
p_list <- lapply(fonts, function(f){
ggseqlogo(seqs_dna$MA0001.1,font=f)+ggtitle(f)
})
do.call(gridExtra::grid.arrange,c(p_list, ncol=4))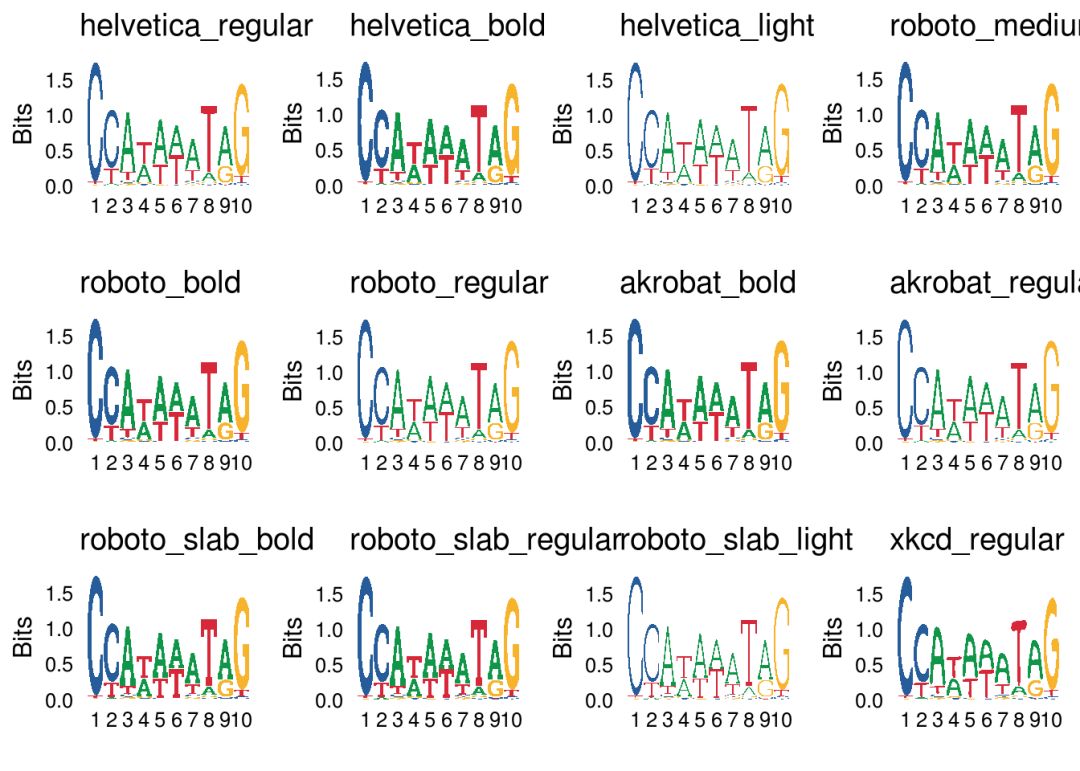
注释
注释的话跟ggplot2是一样的
ggplot()+
annotate("rect", xmin = 0.5, xmax = 3.5, ymin = -0.05, ymax = 1.9, alpha=0.1, col="black", fill="yellow")+
geom_logo(seqs_dna$MA0001.1, stack_width = 0.9)+
annotate("segment", x=4, xend = 8, y=1.2, yend = 1.2, size=2)+
annotate("text", x=6, y=1.3, label="Text annotation")+
theme_logo()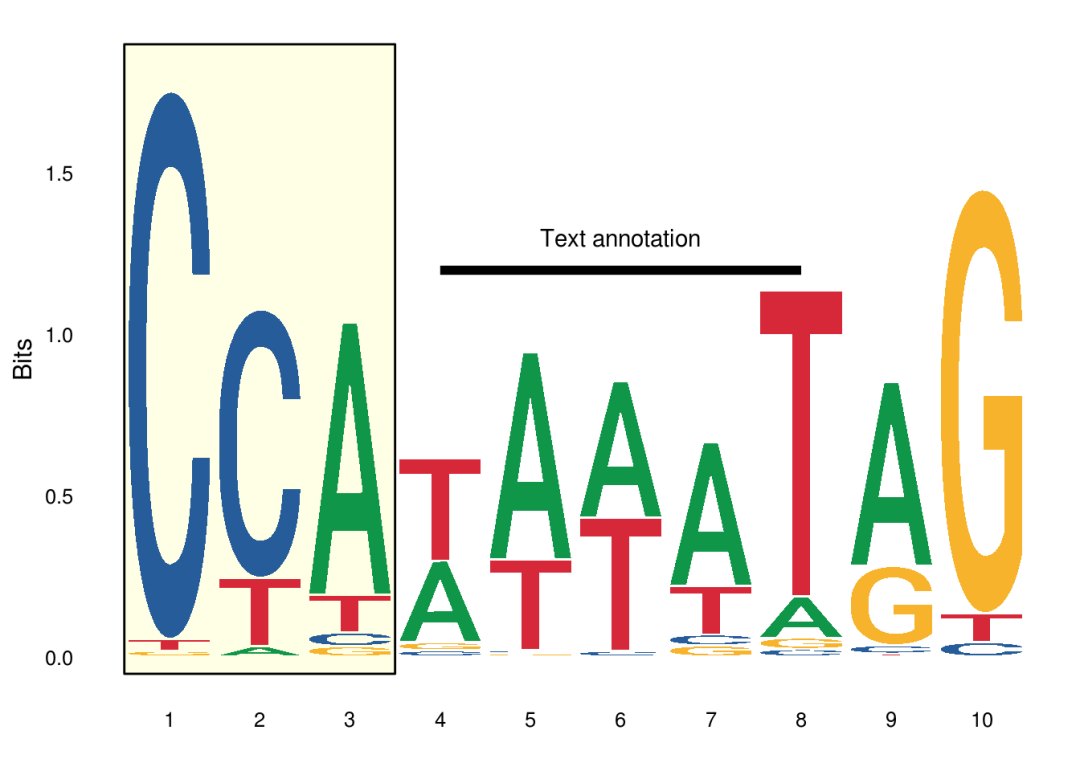
图形组合
将ggseqlogo生成的图形与ggplot2生成的图形组合在一起。
p1 <- ggseqlogo(seqs_dna$MA0008.1)+theme(axis.text.x = element_blank())
aln <- data.frame(
letter=strsplit("AGATAAGATGATAAAAAGATAAGA", "")[[1]],
species=rep(c("a","b","c"), each=8),
x=rep(1:8,3)
)
aln$mut <- "no"
aln$mut[c(2,15,20,23)]="yes"
p2 <- ggplot(aln, aes(x, species)) +
geom_text(aes(label=letter, color=mut, size=mut)) +
scale_x_continuous(breaks=1:10, expand = c(0.105, 0)) + xlab('') +
scale_color_manual(values=c('black', 'red')) +
scale_size_manual(values=c(5, 6)) +
theme_logo() +
theme(legend.position = 'none', axis.text.x = element_blank())
bp_data <- data.frame(
x=1:8,
conservation=sample(1:100, 8)
)
p3 <- ggplot(bp_data, aes(x, conservation))+
geom_bar(stat = "identity", fill="grey")+
theme_logo()+
scale_x_continuous(breaks = 1:10, expand = c(0.105, 0))+
xlab("")
suppressMessages(require(cowplot))
plot_grid(p1,p2,p3,ncol = 1, align = "v")
严涛:浙江大学,作物遗传育种在读博士。爱鼓捣各种可视化软件,爱折腾。点击阅读原文跳转其博客。
更多数据可视化可参考在线工具:http://www.ehbio.com/ImageGP/
(点击图片直达网站手册)
你可能还想看
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集