
【行业资讯】AI改变化学领域,机器学习范式加速化学物质发现
预计阅读时间:12分钟

随着人工智能技术兴起,在化学领域,传统的基于实验和物理模型的方式逐渐与基于数据的机器学习范式融合。越来越多的用于计算机处理数据表示被开发出来,并不断适应着以生成式为主的统计模型。

实验室中的自动化工作。我们可以使用机器学习技术来加速合成工作流程。一种方法使用 “自动驾驶实验室” 来自动化日常任务、优化资源支出并节省时间。一个相对较新的,但值得注意的案例是使用机器人平台 Ada 来自动化薄膜材料的合成、处理和特征化(请参阅此处的平台)。另一项研究展示了使用移动机器人化学家能够操作仪器,并在八天内对 688 次实验进行测量;
化学反应预测。我们可以使用分类模型来预测将发生的反应类型,或者简化问题并预测某个化学反应是否会发生。这个问题有很多不同的建模方法;
化学数据挖掘。像许多其他学科一样,化学有大量可用于研究趋势和相关性的科学文献。一个值得注意的例子是对人类基因组计划提供的大量信息进行数据挖掘,以识别基因组数据的趋势。
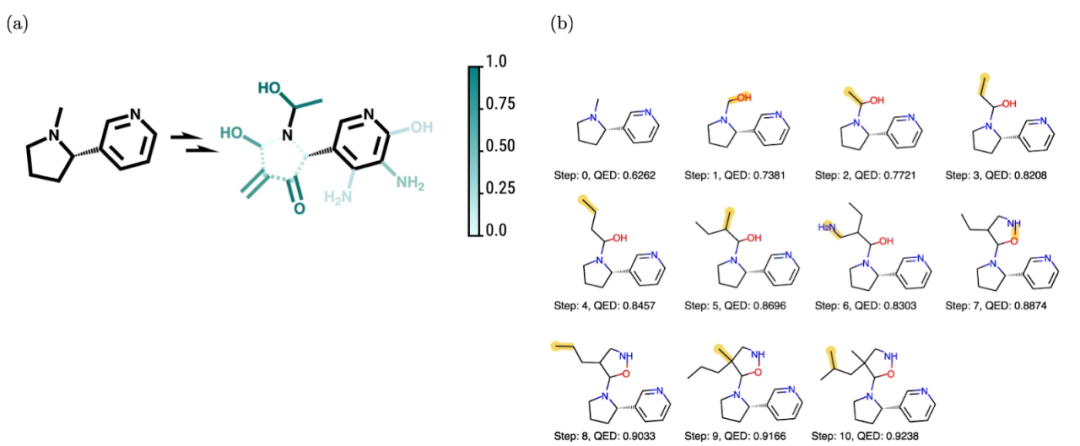
计算和实验之间的差距。虽然计算方法的目标是帮助实现实验的目标,但前者的结果并不总是可以迁移到后者。例如,在使用机器学习寻找候选分子时,我们必须牢记分子在其合成途径中很少是独一无二的,而且通常很难知道未经探索的化学反应是否会在实践中起作用。即便可以起作用,目标化合物的收率、纯度和分离也存在问题。计算工作和实验工作之间的差距甚至会变得更大,因为计算方法所采用的指标并不总是可以转移到后者(上面提到的 QED 只是众多例子中的一个)上,而且实验验证可能不可行;
需要更好的数据库和缺乏基准。由于整个化学空间是无限的,所以我们最希望有足够大的样本量来帮助我们进行之后的泛化。然而,目前大多数数据库都是为不同目的而设计的,它们通常使用不同的文件格式;其中一些缺乏提交的验证程序,或者它们在设计时没有考虑到人工智能的任务。此外,我们拥有的大多数数据库的化学数据范围有限 —— 它们只包含某些类型的分子。最后,大多数涉及使用人工智能进行化学预测的任务都缺乏一个基准平台,这使得许多不同研究的比较变得不可行。AlphaFold 成功的主要原因之一是它提供了上述所有内容作为蛋白质结构预测 (CASP) 竞赛的关键评估的一部分,这表明需要有组织的努力来简化和改进涉及化学预测的其他任务。
THE END
来源 | 机器之心
版权声明:本号内容部分来自互联网,转载请注明原文链接和作者,如有侵权或出处有误请和我们联系。