
Salmon构建索引的时间效率和计算效率明显高于STAR
前面我们评估了不同大小基因组基于STAR构建索引所需的计算资源和时间资源、不同大小数据集基于STAR进行比对所需的计算资源和时间资源和STAR比对速度与分配线程的关系。
将人类基因组按染色体拆分模拟不同大小基因组构建索引的计算资源需求
采用染色体累加的方式,不断模拟不同大小的基因组对计算资源的需求。
gffread GRCh38.gtf -g GRCh38.fa -w GRCh38.transcript.fa.tmp
# gffread生成的fasta文件同时包含基因名字和转录本名字
grep '>' GRCh38.transcript.fa.tmp | head
# 去掉空格后面的字符串,保证cDNA文件中fasta序列的名字简洁,不然后续会出错
cut -f 1 -d ' ' GRCh38.transcript.fa.tmp >GRCh38.transcript.fa
/bin/rm -f GRCh38_tmp.seqid
# i=2
for i in `seq 1 22`; do
echo "chr$i" >>GRCh38_tmp.seqid
seqkit grep -f GRCh38_tmp.seqid GRCh38.fa -o GRCh38_tmp.fa
# 一定得删掉,不然提取转录本信息会少
/bin/rm -f GRCh38_tmp.fa.fai
awk 'BEGIN{OFS=FS="\t"}ARGIND==1{id1[$1]=1}ARGIND==2{if(id1[$1]==1) print $0}' GRCh38_tmp.seqid GRCh38.gtf >GRCh38_tmp.gtf
gffread GRCh38_tmp.gtf -g GRCh38_tmp.fa -w GRCh38.transcript.fa.tmp
grep -c '>' GRCh38.transcript.fa.tmp
# 去掉空格后面的字符串,保证cDNA文件中fasta序列的名字简洁,不然后续会出错
cut -f 1 -d ' ' GRCh38.transcript.fa.tmp >GRCh38.transcript_tmp.fa
grep '^>' GRCh38_tmp.fa | cut -d ' ' -f 1 | sed 's/^>//g' >GRCh38.decoys.txt
# 合并cDNA和基因组序列一起
# 注意:cDNA在前,基因组在后
cat GRCh38.transcript_tmp.fa GRCh38_tmp.fa >GRCh38_trans_genome.fa
# 构建索引 (更慢,结果会更准) # --no-version-check
/bin/rm -rf GRCh38.salmon_sa_index_tmp
/usr/bin/time -o Salmon_human_genome_${i}.log -v salmon index -t GRCh38_trans_genome.fa -d GRCh38.decoys.txt -i GRCh38.salmon_sa_index_tmp -p 2
du -s GRCh38_trans_genome.fa | awk 'BEGIN{OFS="\t"}{print "Genome_size: "$1/10^6}' >>Salmon_human_genome_${i}.log
du -s GRCh38.transcript_tmp.fa | awk 'BEGIN{OFS="\t"}{print "Transcript_size: "$1/10^6}' >>Salmon_human_genome_${i}.log
du -s GRCh38.salmon_sa_index_tmp | awk 'BEGIN{OFS="\t"}{print "Output_size: "$1/10^6}' >>Salmon_human_genome_${i}.log
grep -c '>' GRCh38.transcript_tmp.fa | awk 'BEGIN{OFS="\t"}{print "Transcript_count: "$1}' >>Salmon_human_genome_${i}.log
done
# 获取所有基因组序列的名字存储于decoy中
grep '^>' GRCh38.fa | cut -d ' ' -f 1 | sed 's/^>//g' >GRCh38.decoys.txt
# 合并cDNA和基因组序列一起
# 注意:cDNA在前,基因组在后
cat GRCh38.transcript.fa GRCh38.fa >GRCh38_trans_genome.fa
# 构建索引 (更慢,结果会更准) # --no-version-check
/bin/rm -rf GRCh38.salmon_sa_index
/usr/bin/time -o Salmon_human_genome.log -v salmon index -t GRCh38_trans_genome.fa -d GRCh38.decoys.txt -i GRCh38.salmon_sa_index -p 2
du -s GRCh38_trans_genome.fa | awk 'BEGIN{OFS="\t"}{print "Genome_size: "$1/10^6}' >>Salmon_human_genome.log
du -s GRCh38.transcript.fa | awk 'BEGIN{OFS="\t"}{print "Transcript_size: "$1/10^6}' >>Salmon_human_genome.log
du -s GRCh38.salmon_sa_index | awk 'BEGIN{OFS="\t"}{print "Output_size: "$1/10^6}' >>Salmon_human_genome.log
grep -c '>' GRCh38.transcript.fa | awk 'BEGIN{OFS="\t"}{print "Transcript_count: "$1}' >>Salmon_human_genome.log整合数据生成最终测试结果数据集
首先写一个awk脚本,整理并转换 CPU 使用率、程序耗时、最大物理内存需求。这个脚本存储为timeIntegrate3.awk。
#!/usr/bin/awk -f
function to_minutes(time_str) {
a=split(time_str,array1,":");
minutes=0;
count=1;
for(i=a;i>=1;--i) {
minutes+=array1[count]*60^(i-2);
count+=1;
}
return minutes;
}
BEGIN{OFS="\t"; FS=": "; }
ARGIND==1{if(FNR==1) header=$0; else datasize=$0;}
ARGIND==2{
if(FNR==1 && outputHeader==1)
print "Time_cost\tMemory_cost\tnCPU\tOutput_size\tTranscript_size\tTranscript_count", header;
if($1~/Elapsed/) {
time_cost=to_minutes($2);
} else if($1~/Maximum resident set size/){
memory_cost=$2/10^6;
} else if($1~/CPU/) {
cpu=($2+0)/100
} else if($1~/Output_size/){
Output_size=$2
} else if($1~/Transcript_size/){
Transcript_size=$2
} else if($1~/Transcript_count/){
Transcript_count=$2
}
}
END{print time_cost, memory_cost, cpu, Output_size, Transcript_size, Transcript_count, datasize}用timeIntegrate3.awk整合评估结果
/bin/rm -f GRCh38_salmon_genome_build.summary
for i in `seq 1 22`; do
awk -v i=${i} 'BEGIN{for(j=1;j<=i;j++) scaffold["chr"j]=1;}\
{if(scaffold[$1]==1) genomeSize+=$2}END{print "Genome_size";print genomeSize/10^9}' star_GRCh38/chrNameLength.txt | \
awk -v outputHeader=$i -f ./timeIntegrate3.awk - Salmon_human_genome_${i}.log >>GRCh38_salmon_genome_build.summary
done
awk '{genomeSize+=$2}END{print "Genome_size"; print genomeSize/10^9}' star_GRCh38/chrNameLength.txt \
| awk -f ./timeIntegrate3.awk - Salmon_human_genome.log >>GRCh38_salmon_genome_build.summary最终获得结果文件如下:
Time_cost Memory_cost nCPU Output_size Transcript_size Transcript_count Genome_size
4.39933 1.39078 2.27 1.27743 0.036424 20716 0.248956
9.0455 2.82505 2.28 2.59288 0.067352 38135 0.49115
12.481 4.01651 2.28 3.66561 0.091868 52968 0.689446
15.8388 5.15384 2.29 4.69 0.109064 62709 0.87966
20.1408 6.3714 2.32 5.78156 0.127944 74002 1.0612
22.9222 8.55484 2.3 6.72696 0.147956 85002 1.232
25.6965 9.34814 2.32 7.61427 0.166756 96223 1.39135
28.2602 10.0071 2.31 8.41734 0.181776 105970 1.53649
30.9775 13.3335 2.32 9.15804 0.196396 113998 1.67488
33.9318 14.3744 2.32 10.096 0.211736 122370 1.80868
38.1807 12.1258 2.33 10.8738 0.23446 136491 1.94377
40.3075 16.167 2.34 11.6486 0.255484 149677 2.07704
42.642 16.8637 2.34 12.2845 0.26292 154002 2.19141
47.5528 16.7611 2.35 12.8888 0.276596 162687 2.29845
47.7633 17.2338 2.36 13.4525 0.291424 171376 2.40044
51.327 17.7096 2.35 13.9687 0.308412 182566 2.49078
54.8048 18.2542 2.39 14.4603 0.329668 196504 2.57404
55.135 18.6732 2.35 14.9188 0.337088 201042 2.65441
56.1127 19.1378 2.37 15.2844 0.357176 214955 2.71303
57.8897 19.4656 2.35 15.6548 0.366436 220525 2.77747
58.7447 19.7024 2.38 15.9044 0.371412 223564 2.82418
59.8983 19.9482 2.36 16.185 0.379824 228615 2.875
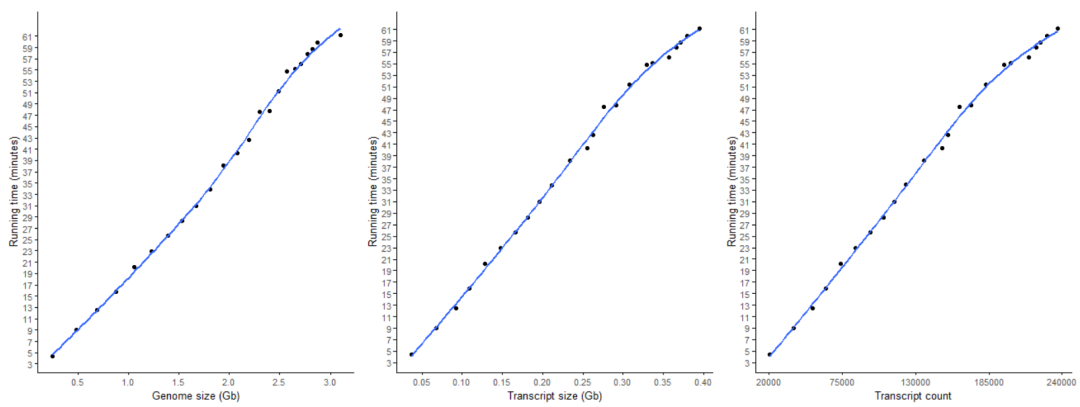
61.1667 21.1396 2.37 17.4117 0.394968 236920 3.09975构建索引的时间随数据量的变化
Salmon构建索引的时间随基因组大小/染色体大小/染色体数目增加而增加,基本成线性关系
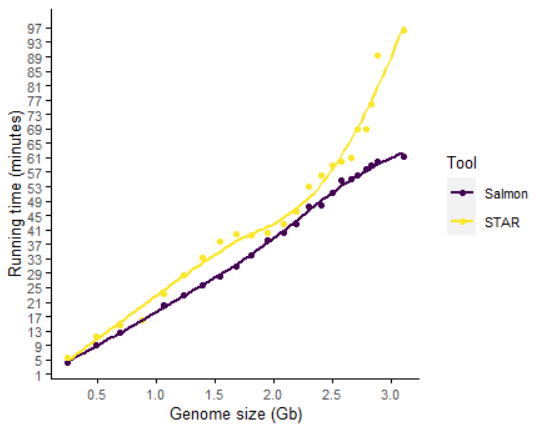
同样基因组大小,给定相同线程数时,Salmon速度快于STAR。
数据量更大时,salmon的速度优势更明显。
library(ImageGP)
library(patchwork)
p1 <- sp_scatterplot("GRCh38_salmon_genome_build.summary", melted = T, xvariable = "Genome_size",
yvariable = "Time_cost", smooth_method = "auto",
x_label ="Genome size (Gb)", y_label = "Running time (minutes)") +
scale_x_continuous(breaks=seq(0,4, by=0.5)) +
scale_y_continuous(breaks=seq(1,62,by=2))
p2 <- sp_scatterplot("GRCh38_salmon_genome_build.summary", melted = T, xvariable = "Transcript_size",
yvariable = "Time_cost", smooth_method = "auto",
x_label ="Transcript size (Gb)", y_label = "Running time (minutes)") +
scale_x_continuous(breaks=seq(0,0.4, by=0.05)) +
scale_y_continuous(breaks=seq(1,62,by=2))
p3 <- sp_scatterplot("GRCh38_salmon_genome_build.summary", melted = T, xvariable = "Transcript_count",
yvariable = "Time_cost", smooth_method = "auto",
x_label ="Transcript count", y_label = "Running time (minutes)") +
scale_x_continuous(breaks=seq(20000,240000, length=5)) +
scale_y_continuous(breaks=seq(1,62,by=2))
p1 + p2 + p3salmon_genome <- sp_readTable("GRCh38_salmon_genome_build.summary")
star_genome <- sp_readTable("GRCh38_star_genome_build.summary")
salmon_genome_merge <- salmon_genome[,c("Time_cost", "Memory_cost", "nCPU", "Output_size", "Genome_size"), drop=F]
salmon_genome_merge$Tool <- "Salmon"
colnames(star_genome) <- c("Time_cost", "Memory_cost", "nCPU", "Genome_size", "Output_size" )
star_genome <- star_genome[,c("Time_cost", "Memory_cost", "nCPU", "Genome_size", "Output_size" )]
star_genome$Tool <- "STAR"
genome_build_merge <- rbind(salmon_genome_merge, star_genome)
sp_scatterplot(genome_build_merge, melted = T, xvariable = "Genome_size",
yvariable = "Time_cost", smooth_method = "auto",
color_variable = "Tool",
x_label ="Genome size (Gb)", y_label = "Running time (minutes)") +
scale_x_continuous(breaks=seq(0,4, by=0.5)) +
scale_y_continuous(breaks=seq(1,100,by=4))构建索引的所需内存随数据量的变化
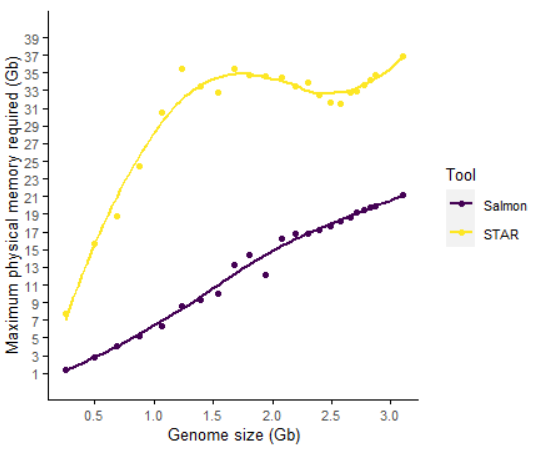
Salmon构建索引的内存需求随基因组大小/染色体大小/染色体数目增加而增加,基本成线性关系

Salmon对内存的需求明显小于STAR的需求。
对人类3G大小的基因组,Salmon的内存需求只占STAR的一半
如果用Salmon构建索引时不考虑基因组信息所需内存更少
p1 <- sp_scatterplot("GRCh38_salmon_genome_build.summary", melted = T, xvariable = "Genome_size",
yvariable = "Memory_cost", smooth_method = "auto",
x_label ="Genome size (Gb)", y_label = "Maximum physical memory required (Gb)") +
scale_x_continuous(breaks=seq(0.5,5, by=0.5)) +
scale_y_continuous(breaks=seq(1,22,length=22))
p2 <- sp_scatterplot("GRCh38_salmon_genome_build.summary", melted = T, xvariable = "Transcript_size",
yvariable = "Memory_cost", smooth_method = "auto",
x_label ="Transcript size (Gb)", y_label = "Maximum physical memory required (Gb)") +
scale_x_continuous(breaks=seq(0,0.4, by=0.05)) +
scale_y_continuous(breaks=seq(1,22,length=22))
p3 <- sp_scatterplot("GRCh38_salmon_genome_build.summary", melted = T, xvariable = "Transcript_count",
yvariable = "Memory_cost", smooth_method = "auto",
x_label ="Transcript count", y_label = "Maximum physical memory required (Gb)") +
scale_x_continuous(breaks=seq(20000,240000, length=5)) +
scale_y_continuous(breaks=seq(1,22,length=22))
p1 + p2 + p3
sp_scatterplot(genome_build_merge, melted = T, xvariable = "Genome_size",
yvariable = "Memory_cost", smooth_method = "auto",
color_variable = "Tool",
x_label ="Genome size (Gb)", y_label = "Maximum physical memory required (Gb)") +
scale_x_continuous(breaks=seq(0,4, by=0.5)) +
scale_y_continuous(breaks=seq(1,40,by=2),limits=c(0,40))构建索引时对 CPU 的利用率
Salmon的CPU利用率跟数据大小关系不大,且并行效率很高。
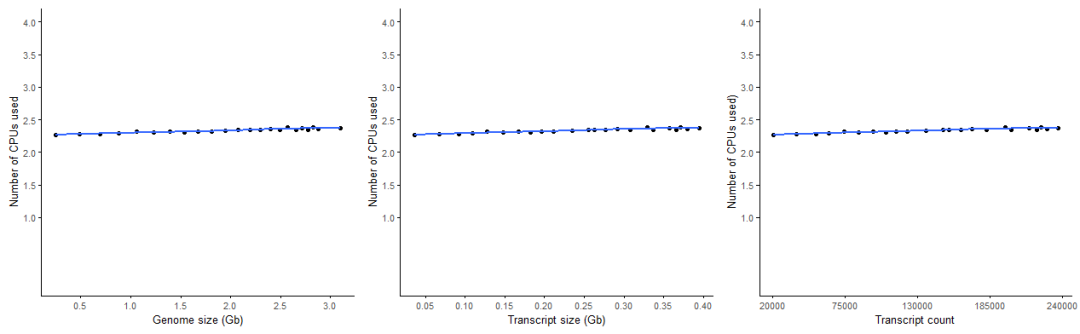
Salmon的CPU利用率更稳定,且明显高于STAR

p1 <- sp_scatterplot("GRCh38_salmon_genome_build.summary", melted = T, xvariable = "Genome_size",
yvariable = "nCPU", smooth_method = "auto",
x_label ="Genome size (Gb)", y_label = "Number of CPUs used") +
scale_x_continuous(breaks=seq(0.5,5, by=0.5)) +
scale_y_continuous(breaks=seq(1,4.5,by=0.5), limits=c(0,4))
p2 <- sp_scatterplot("GRCh38_salmon_genome_build.summary", melted = T, xvariable = "Transcript_size",
yvariable = "nCPU", smooth_method = "auto",
x_label ="Transcript size (Gb)", y_label = "Number of CPUs used") +
scale_x_continuous(breaks=seq(0,0.4, by=0.05)) +
scale_y_continuous(breaks=seq(1,4.5,by=0.5), limits=c(0,4))
p3 <- sp_scatterplot("GRCh38_salmon_genome_build.summary", melted = T, xvariable = "Transcript_count",
yvariable = "nCPU", smooth_method = "auto",
x_label ="Transcript count", y_label = "Number of CPUs used)") +
scale_x_continuous(breaks=seq(20000,240000, length=5)) +
scale_y_continuous(breaks=seq(1,4.5,by=0.5), limits=c(0,4))
p1 + p2 + p3sp_scatterplot(genome_build_merge, melted = T, xvariable = "Genome_size",
yvariable = "nCPU", smooth_method = "auto",
color_variable = "Tool",
x_label ="Genome size (Gb)", y_label = "Number of CPUs used)") +
scale_x_continuous(breaks=seq(0,4, by=0.5)) +
scale_y_continuous(breaks=seq(0,3,by=0.5),limits=c(0,3))生成的索引大小根基因组大小的关系 (磁盘需求)
Salmon生成的索引大小跟基因组大小正相关

Salmon构建的索引占用磁盘空间更小
基因组增大时,Salmon所需磁盘空间增速小于STAR

p1 <- sp_scatterplot("GRCh38_salmon_genome_build.summary", melted = T, xvariable = "Genome_size",
yvariable = "Output_size", smooth_method = "auto",
x_label ="Genome size (Gb)", y_label = "Disk space usages (Gb)") +
scale_x_continuous(breaks=seq(0.5,5, by=0.5)) +
scale_y_continuous(breaks=seq(1,18,by=1))
p2 <- sp_scatterplot("GRCh38_salmon_genome_build.summary", melted = T, xvariable = "Transcript_size",
yvariable = "Output_size", smooth_method = "auto",
x_label ="Transcript size (Gb)", y_label = "Disk space usages (Gb)") +
scale_x_continuous(breaks=seq(0,0.4, by=0.05)) +
scale_y_continuous(breaks=seq(1,18,by=1))
p3 <- sp_scatterplot("GRCh38_salmon_genome_build.summary", melted = T, xvariable = "Transcript_count",
yvariable = "Output_size", smooth_method = "auto",
x_label ="Transcript count", y_label = "Disk space usages (Gb)") +
scale_x_continuous(breaks=seq(20000,240000, length=5)) +
scale_y_continuous(breaks=seq(1,18,by=1))
p1 + p2 + p3sp_scatterplot(genome_build_merge, melted = T, xvariable = "Genome_size",
yvariable = "Output_size", smooth_method = "auto",
color_variable = "Tool",
x_label ="Genome size (Gb)", y_label = "Disk space usages (Gb)") +
scale_x_continuous(breaks=seq(0,4, by=0.5)) +
scale_y_continuous(breaks=seq(0,30,by=2),limits=c(0,30))往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集

评论