
哈佛、哥大开源1600万组蛋白质序列,解决AlphaFold 2训练数据私有难题!

新智元报道
新智元报道
编辑:Aeneas
【新智元导读】这个叫OpenProteinSet的开源数据集,提供1600万个开源蛋白质多序列对齐(MSA)和相关数据。它涵盖了所有蛋白质数据库的蛋白质和各种UniProt集群,可以实现广泛的AI模型训练。
蛋白质是生命的主力军,了解它们的序列和结构,是设计新酶、开发救命药物等生物学和医学挑战的关键。
DeepMind的AlphaFold 2,能够以前所未有的准确性预测蛋白质结构。
然而,由于缺乏开放的训练数据,这一领域的进展被严重阻碍。
但来自哈佛大学、哈佛医学院、哥伦比亚大学、纽约大学和Flatiron Institute的研究者,引入了一个开源数据库。
这个名为OpenProteinSet的开源数据库,可以通过大规模提供蛋白质比对数据,来大大改善这种状况。
它提供的数据集,和用于训练AlphaFold 2的数据集质量相同。

因为AlphaFold 2,MSA的实用性爆炸性增长
蛋白质的功能,就编码在氨基酸序列中。
在进化过程中,这些序列会积累一些微小的变化,而蛋白质的整体结构和功能却一直保持不变。
多序列对齐(MSA)是一组和进化相关的蛋白质序列,通过插入间隙进行对齐,使匹配的氨基酸最终出现在同一列中。
通过分析这些MSA中的模式,可以深入了解蛋白质的结构和功能。
MSA的每一行,都是一个蛋白质序列。蛋白质是由20个氨基酸(或「残基」)组成的一维字符串,每个氨基酸或「残基」由一个字母表示。
目标或「查询」 蛋白质在MSA的第一行中给出。后续行是根据与查询序列的相似性,从大型序列数据库中检索到的进化相关(「同源」) 蛋白质。
为了改进比对、适应长度随时间变化的同源序列,MSA比对软件可以在同源序列中插入「缺口」(此处用破折号表示)或删除残基。
MSA中同源序列的数量(「深度」)及其多样性,都有助于MSA的实用性。
 MSA引物
MSA引物
长期以来,MSA对蛋白质研究都至关重要,不过在2021年,因为AlphaFold 2的出现,MSA的实用性呈现了爆炸性增长。
通过MSA,AlphaFold 2能够以近乎实验级的准确性预测蛋白质结构。
然而有一个问题:虽然AlphaFold 2是开源的,但它的训练数据仍然是私有的。
这样做的计算成本很高。根据目标序列长度和正在搜索的序列数据库的大小生成一个具有高灵敏度的MSA,可能需要几个小时。
这样,蛋白质机器学习和生物信息学的前沿研究除了少数大型研究团队外,其他所有人都无法访问。
1600万个MSA全部开源
因此,团队提出了OpenProteinSet,这是一个在AlphaFold 2及其以上规模训练生物信息学的模型。
它包含了AlphaFold 2未发布的训练集,包括所有唯一的蛋白质数据库(PDB)链的MSAs和结构模板。
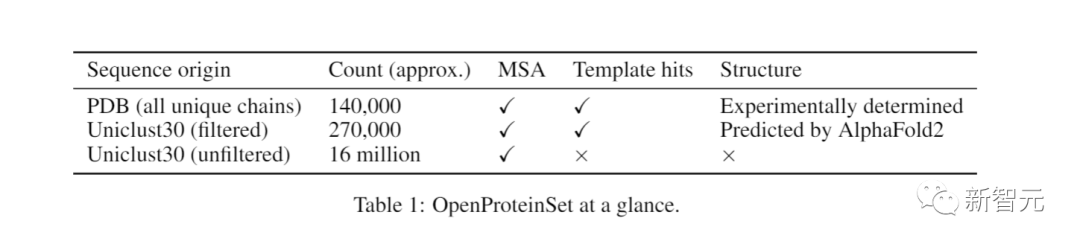
现在,OpenProteinSet提供了1600万个MSA和相关数据,并且全部开源。
PDB是实验确定的蛋白质结构的权威数据库,而OpenProteinSet包括PDB中所有140,000种蛋白质的MSA。
它甚至还包括来自UniProt知识库的序列,该序列按相似性聚类。
对于PDB蛋白质,OpenProteinSet能够提供来自多个序列数据库的原始MSA。
通过搜索PDB,它还能找到结构相似的蛋白质。
AlphaFold 2预测的结构,包括270,000个不同的UniProt集群。
使用开源数据集重新创建AlphaFold 2
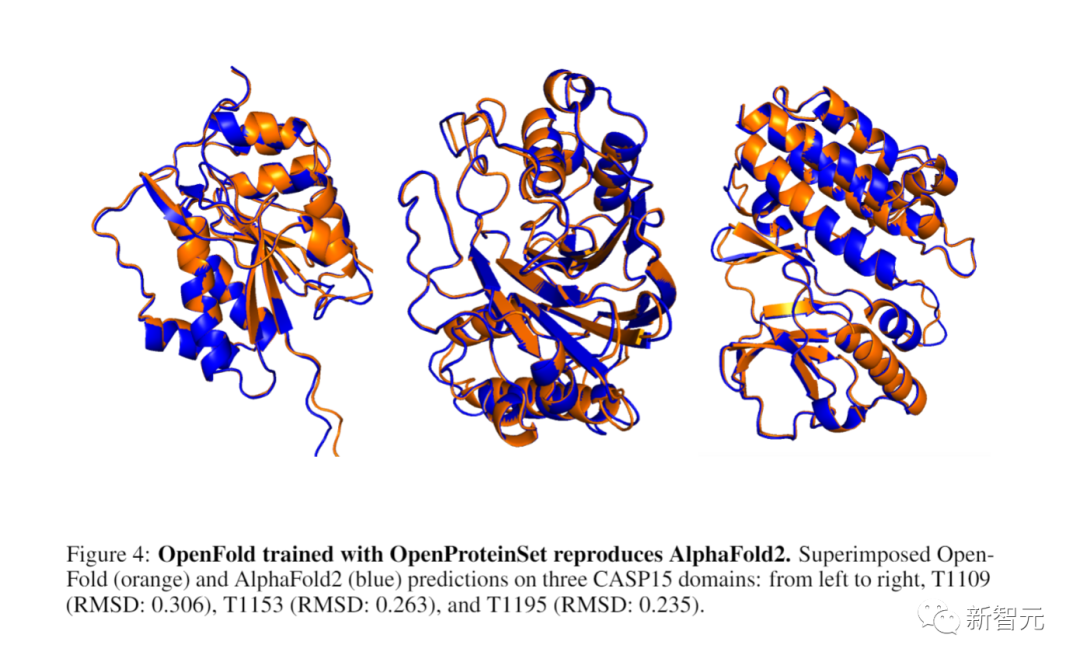
开发者还会使用OpenProteinSet来训练OpenFold,这是AlphaFold 2的一个开放版本。
他们发现,OpenFold的性能与DeepMind的原始数据相当,证明了这种开放数据的充分性。
团队表示,「通过OpenProteinSet,我们大大提高了分子机器学习社区可用的预计算MSA的数量和质量,」
该数据集可直接应用于结构生物学的各种任务。
实验方法
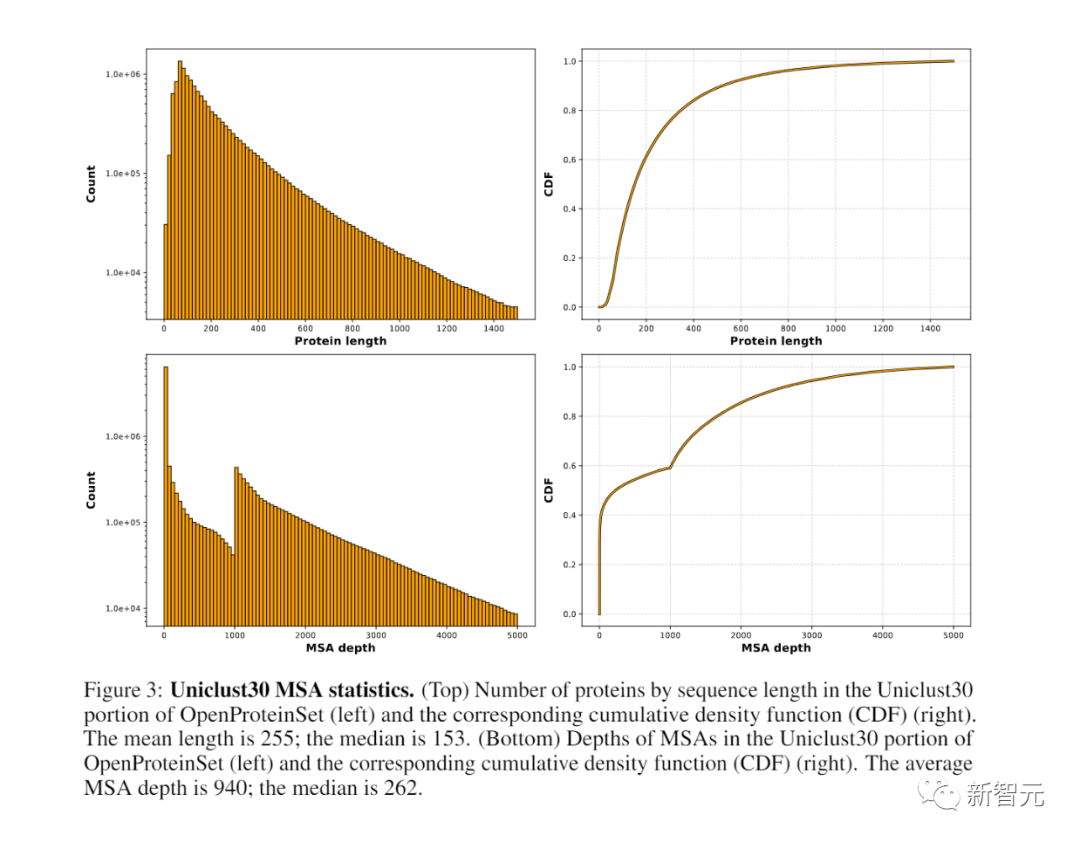
OpenProteinSet由超过1600万个独特的MSAs组成,这些MSAs是根据AIphaFold2论文中的程序生成的。
这一计数包括截至2022年4月PDB中所有14万个唯一链的MSAs,以及针对同一数据库为Uniclust30中的每个序列集群计算的1,600万个MSAs。
从后一组中,研究者确定了270,000个最大多样性代表性集群,比如可以适用于AphaFold2训练过程中的自我蒸馏集。
对于每个PDB链,研究者使用了不同的对齐工具和序列数据库计算三个MSAs。
使用OpenFold中的脚本,可以从公开可用的PDBmmCIF文件中,检索相应的结构。
与用于生成AIphaFold2训练集的过程一样,研究者更改了MSA生成工具的一些默认选项。
随后,产生了大约1600万个MSAs,每个集群一个。
为了创建一个不同的、深度的MSAs子集,研究者通过迭代去除代表性链出现在其他MSAs中最多的MSAs。
这样重复,直到每个代表链只出现在它自己的MSA中。
为了与对应的(未发布的)AlphaFold 2集进行奇偶性检验,研究者进一步删除了代表序列大于1024个残基或小于200个残基的簇。
最后,他们剔除了相应MSAs少于200个序列的簇,只剩下270,262个MSAs。
总的来说,OpenProteinSet中的MSAs代表了超过400万小时的计算。
OpenProteinSet大大提高了分子机器学习社区可用的预计算MSAs的数量和质量,它可以直接应用于结构生物学中的各种任务。
随着模型对数据的需求越来越大,像OpenProteimnSet这样的数据库既可以作为多模态语言模型的生物知识宝库,也可以作为多模态训练本身的实证研究工具。
总之,OpenProteinSet将进一步推动生物信息学、蛋白质机器学习等领域的研究。
参考资料:
https://the-decoder.com/openproteinset-provides-open-source-training-data-for-structural-biology-at-scale/
https://arxiv.org/pdf/2308.05326.pdf


