
iMeta | 南科大夏雨组纳米孔测序揭示微生物可减轻高海拔冻土温室气体排放
基于纳米孔测序的宏基因组学揭示微生物作为生物过滤器减轻高海拔冻土的温室气体排放

https://doi.org/10.1002/imt2.24
Research Article
Volume1, Issue2

●2022年5月8日,南方科技大学夏雨团队在iMeta在线发表了题为“Microorganisms as bio-filters to mitigate greenhouse gas emissions from high-altitude permafrost revealed by nanopore-based metagenomics”的方法类文章。
● 该文章使用纳米孔宏基因组测序和Illumina高通量RNA-seq技术,探究了祁连山冻土活动层冻融循环中的微生物功能活性,发现甲基单胞菌可以作为生物过滤器来减轻冻土融化过程的甲烷排放。
● 第一作者:党晨原
● 通讯作者:夏雨 (xiay@sustech.edu.cn)
● 合作作者:吴子麒,张淼,李响,孙雨芹,吴仁安,郑焰
青藏高原独特的气候和地理条件使高海拔冻土的退化比极地更为严重。然而,冻土融化中和温室气体产生相关的微生物响应仍不十分清楚。本研究使用纳米孔宏基因组测序和高通量RNA-seq技术,探究了祁连山冻土活动层冻融循环中的微生物功能活性。本研究建立了一个生物信息学分析流程促进非组装纳米孔宏基因组的系统发育和功能的注释。通过使用该策略,冻土中属水平物种注释量增加了42%,氮和甲烷循环的相关基因注释量增加了58%。基于上述结果,我们观察到甲基单胞菌(Methylomonas)强烈的甲烷有氧氧化作用,其可以作为生物过滤器来减轻冻土融化过程的甲烷排放。尽管在冻土融化过程中乙酸型产甲烷途径的转录活性明显增强,但产生的甲烷可被微生物有效地消耗。通过现场气相测量和培养实验发现二氧化碳是冻土释放碳的主要形式,进一步证实了上述的过滤作用。此外,冻土中的氮代谢趋于闭路循环,并且也在现场检测到了近地表的氧化亚氮被表层冻土中的微生物群落消耗。我们的研究表明,虽然冻土融化状态的增加促进了异养氮和甲烷代谢,但其中活跃的微生物可作为生物过滤器来减少冻土融化释放的温室气体。
关键词:移码校正;全球变暖;高海拔冻土;宏转录组学;纳米孔测序
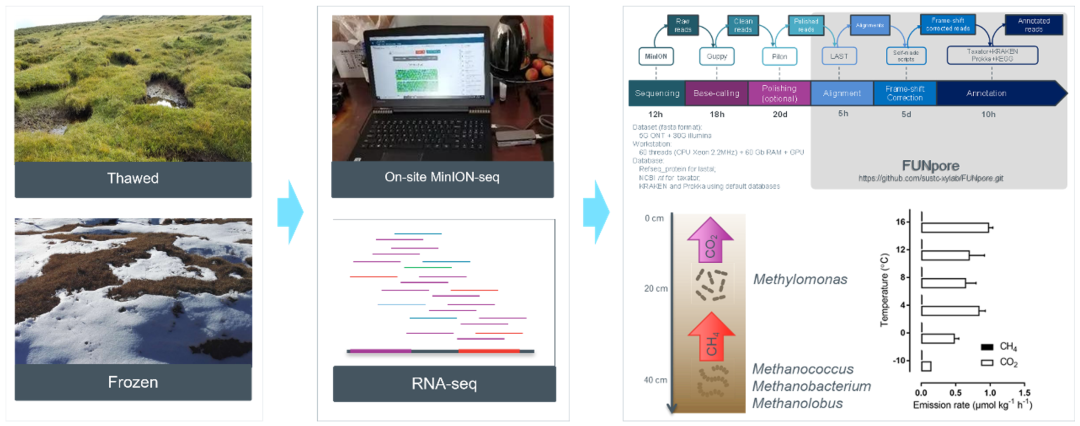
● 使用基于Oxford Nanopore Technologies(ONT)的现场宏基因组测序和基于Illumina RNA-seq的宏转录组测序技术,探究了祁连山多年冻土活动层中的微生物功能
● 使用自主开发的ONT数据校正注释工具(FUNpore),检测到的属和基因分别增加了42%和58%
● 甲基单胞菌的好氧氧化作用可作为生物过滤器来减轻冻土的甲烷排放
● 硝酸盐异化还原为铵(DNRA)途径促进了冻土中氮的闭路循环,并在现场冻土表层观察到了 N2O的消耗
Bilibili:https://www.bilibili.com/video/BV1y5411Q7Ya/
Youtube:https://youtu.be/NiySWnBBqFI
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
引 言
地球表面约有25%被永久冻土覆盖,约含有1672 Pg碳,大致相当于植被和大气中的碳总量。暴露于季节性冻融循环的永久冻土表层被称为活土层,在近几十年来全球变暖的背景下,活土层厚度逐渐增加,下层的永久冻土随之减少,有些地方的冻土甚至已经消失。随着永久冻土的融化,预计到2100年,近地表(地表以下3米以内)大约有174 Pg的碳可被微生物降解。由于全球变暖,大量被封存的碳可能被复苏的微生物降解并导致温室气体排放。
近年来,对永久冻土中参与有机碳代谢的微生物群落、功能和活动的研究越来越多。之前的研究表明,尽管永久冻土条件恶劣,仍含有多种多样的微生物群落。这些微生物不仅能生存、生长,还能参与代谢活动,说明这些微生物已经适应了永久冻土的生物物理环境。最近的研究也揭示了在冻土活动层融冻循环状态下潜在活性细菌群落的变化。此外,在冻土融化过程中,微生物的组成和代谢活动反应迅速,尤其是参与碳和氮代谢的基因,它们在有机质的降解中起着非常重要的作用。三种主要的温室气体,甲烷(CH4)、氧化亚氮(N2O)和二氧化碳(CO2)都与微生物碳、氮代谢直接相关。然而,当前大多数关于永久冻土微生物代谢活性的研究都是在极地地区进行的。
青藏高原是除北极和南极之外最大的冰冻圈,也具有世界上最大的高海拔永久冻土层。由于地质特征较年轻、地温梯度较大,青藏高原的永久冻土比极地地区的薄且温度高。此外,强烈的太阳辐射、稀缺的有机层以及区域性更强的大气变暖使青藏高原地区的高海拔永久冻土比高纬度极低地区的退化更严重。而不同气候和地理条件引起的高海拔永久冻土层融化会影响微生物群落及其功能。然而,尽管已经有研究关注了青藏高原高海拔永久冻土中微生物群落(主要使用基于扩增的方法),但全球变暖对该地区微生物功能组成和代谢活性的影响尚不清楚。
本研究使用基于Oxford Nanopore Technologies(ONT)的宏基因组和基于Illumina的RNA-seq技术,以青藏高原祁连山地区的3000、3500和4000米三个不同海拔高度的永久冻土活动层为研究对象,比较了融化期(8月采样)和冻结期(11月采样)的3500米冻土活动层的微生物代谢活动变化。以期达到以下目的:(1)提供一个新的生物信息学流程(FUNpore)来分析大量的非组装的冻土宏基因组数据,以发挥ONT长读长测序的优势;(2)揭示高海拔冻土活动层在冷冻和融化状态下与温室气体产生相关的微生物活性变化特点。
结果与讨论
使用FUNpore pipeline进行ONT序列校正和功能预测
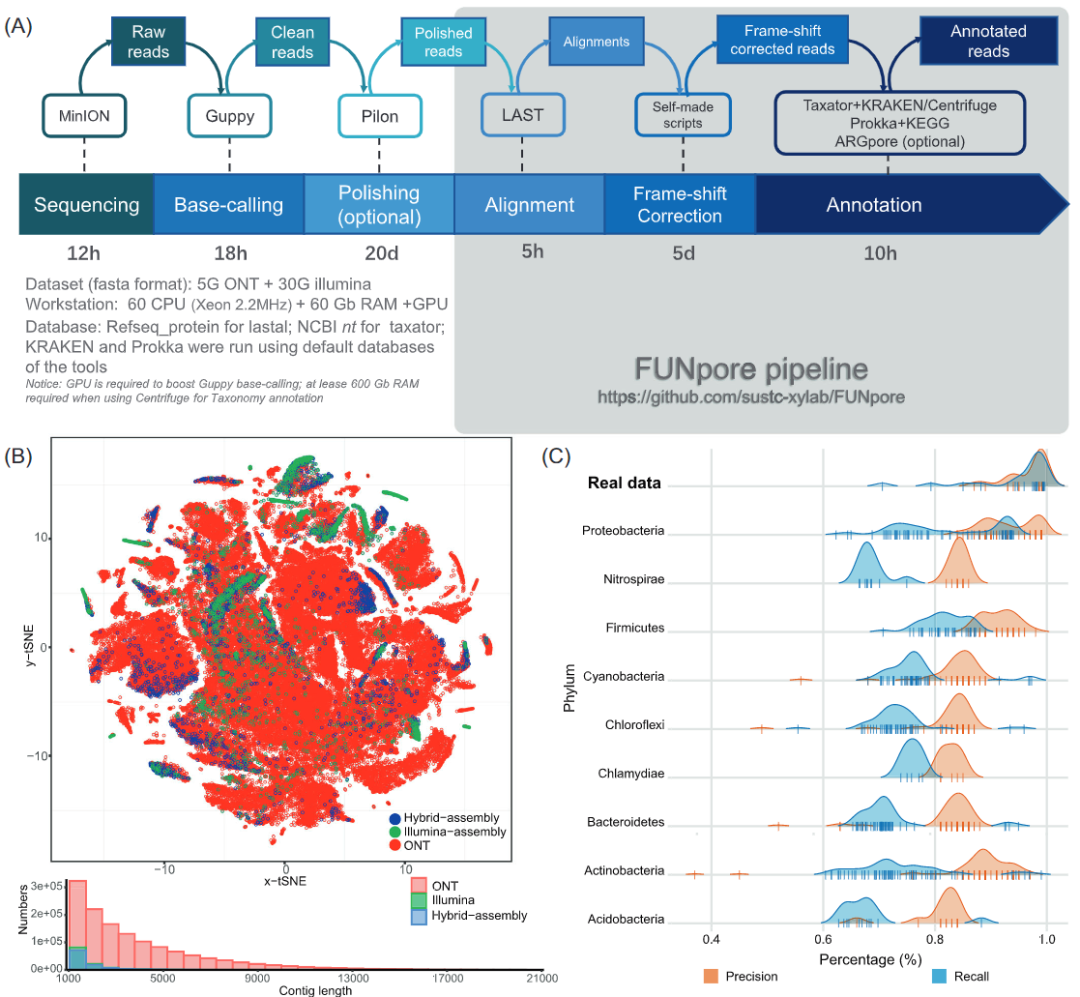
图1 A.FUNpore基于校正的注释工作流程。B.ONT long-read、Illumina数据组装和混合组装(Illumina+ONT)的contigs的五碱基频率的t-SNE分析。C.由真实和模拟的ONT数据评估FUNpore 功能注释的精度和回收率。
虽然ONT读长较长可以显著降低分离菌株和ZymoBIOMICS模拟群落基因组的组装水平,但对于宏基因组数据集,无论是单独组装ONT reads,还是用高质量Illumina短reads混合ONT reads组装,在reads利用率和组装连续性方面都没有很好的效果(图1B)。ONT数据集携带的基因组信息中只有很少一部分能够被整合到最终的contig中(图1B),此外,混合组装过程中消耗巨大的计算资源(主要在服务器RAM上)。对于永久冻土中的微生物群落,使用metaSPAdes和Opera-MS软件来组装5.2 Gb ONT数据(平均读长为3.8 kb)和42 Gb双端Illumina数据(2×150 bp)。与单独使用Illumina数据组装结果相比,混合组装几乎没有改善contig的连续性(图1B,表S5),67.4%的ONT数据并没有在混合装配中使用(表S5)。因为短Illumina reads无法映射低质量区域,ONT reads上随机分布的低质量区域实际上都被组装算法切割开了,所以目前的混合组装软件并没有识别到长ONT reads的长噪声信号(图S2)。这些结果导致ONT long-reads被切割成碎片,在混合组装过程中损失了long-read的优点,类似于仅由short-read组装的contigs。此外,专门为ONT long-read设计的组装工具,如Miniasm和Wtdbg2,也面临着宏基因组数据集中大量数据的丢失问题。使用5.2 Gb的永久冻土ONT数据中只获得了大约5.3 Mb和28.0 Mb的基因组信息(表S5),主要原因是宏基因组数据集内的高多样性导致普遍缺乏足够的覆盖度,这个问题较难通过改进算法来解决。
因此,为了尽量保留ONT数据所携带的基因组信息,本研究的宏基因组分析采用了另一种日益流行的方法:基于错误基因修正的注释。首先,将质量过低(Qscore低于5)的碱基替换为N,避免干扰后续的校正。接下来通过基于Pilon对大量NGS短reads和纳米孔reads的比对,对SNP和Indel进行校正。基于主流映射工具,包括bwa、bbmap、minimap2和LAST的映射灵敏度和运行时间之间的选择,最终使用“mem”参数的bwa能显示出最佳性能(表S6)。根据我们对ONT数据的15000个reads子集的评估,在Pilon抛光后,平均基础精度从94%提高到96%(图S3)。然而,抛光大于50%碱基的纳米孔reads只有59.01%,抛光大于80%碱基的纳米孔reads只有29.40%(图S3),这表明很大一部分的纳米孔reads由于存在无法映射的低质量区域而无法抛光(图S2)。因此最后一步,我们根据LAST的移码比对DNA-蛋白比对,结合高质量的NCBI Refseq_protein数据库,进一步校正了由ONT reads上的剩余错误引起的可区分的移码错误。受到MEGAN6基于DIAMOND的移码比对的校正算法的启发,我们开发了自己的基于LAST对齐的移码校正脚本。由于LAST比对比DIAMOND长得多,因此FUNpore可以校正更多的纳米孔reads移码错误。我们观察到FUNpore平均每千字节可发现27.0移码错误(表S3),而MEGAN6平均每千字节只发现14.8移码错误。
我们评估了基于校正后ONT reads的功能预测精度,尽管发现了大量的假基因,但基于校正后的ONT数据的功能预测显示出了与黄金标准混合组装的高度一致性(平均精度为96.6%,回收率为94.5%)(图1C)。然而对于模拟数据集的性能稍差(精度在80%到94%之间,回收率在65%到97%之间),主要是因为模拟数据集中随机引入的错误的分布模式与真实的纳米孔reads中的分布模式不同。基于模拟数据集,可以观察到Proteobacteria、Firmicutes和Actinobacteria不同门之间的明显差异(图1C)。具有较高校正效率的门类,应是由于它们在Refseq_protein数据库中具有较高的代表性,所以可以产生更多的比对,便于FUNpore移码校正。为了提高FUNpore框架的可用性,我们还结合Taxator和KRAKEN的结果,在FUNpore框架中加入了一个系统发育注释步骤(图1A)。
在非组装的ONT long-read宏基因组方法的帮助下,我们在前所未有的规模上解释冻土微生物群落的代谢潜力(图1B)。与混合组装相比,我们可以在属的水平上注释的冻土微生物组数量增加了42%(图S4A)。此外,我们的策略将涉及氮和甲烷代谢的KO类别(本研究的主要研究对象)扩大了58%(图S4B)。与之对应,基于非组装的ONT long-read宏基因组数据的代谢网络显示出了更高的氮/甲烷代谢功能群体多样性(图S5)。
不同海拔解冻冻土的微生物群落
首先,我们使用MinION对两个独立的HP4000样品(一个现场测序,一个实验室测序)分别进行测序来评估实验的重复性,在属的水平上比较了这两个样品的群落组成。Pearson相关分析显示R2值为0.99(p值< 2.2e−16,图S6),说明群落结构高度相似,同时说明我们在样品采集、运输过程中的保存和测序处理手段都是可靠的。因此,在垂直高寒地区和冻融地区群落的比较中,不应存在人为引入的偏差。
宏基因组分析显示,永久冻土的核心群落沿海拔梯度是保守的(图2),在所观察到的915个属中,有78.4%(719个)个属是共有的(图S7),共有属的丰富度占每个样品总丰度的99%以上。为进一步研究群落的分化,我们对比了丰富度较高的属(所有样品中的相对丰度>0.5‰),发现其中93.8%的优势属都属于变形菌门(Proteobacteria)和放线菌门(Actinobacteria)。群落分化的三元图(图2A)显示,大多数优势属集中在三元图的中心,表明它们在不同研究海拔的群落中具有相似的丰度。然而,有机碳、氮、磷的含量等物理化学因子沿海拔变化明显(表S1),说明高海拔冻土中的微生物群落对土壤化学成分的变化具有抵抗性。这种稳定的抵抗物化改变的核心群落结构,与之前在高海拔环境下的研究结果一致。
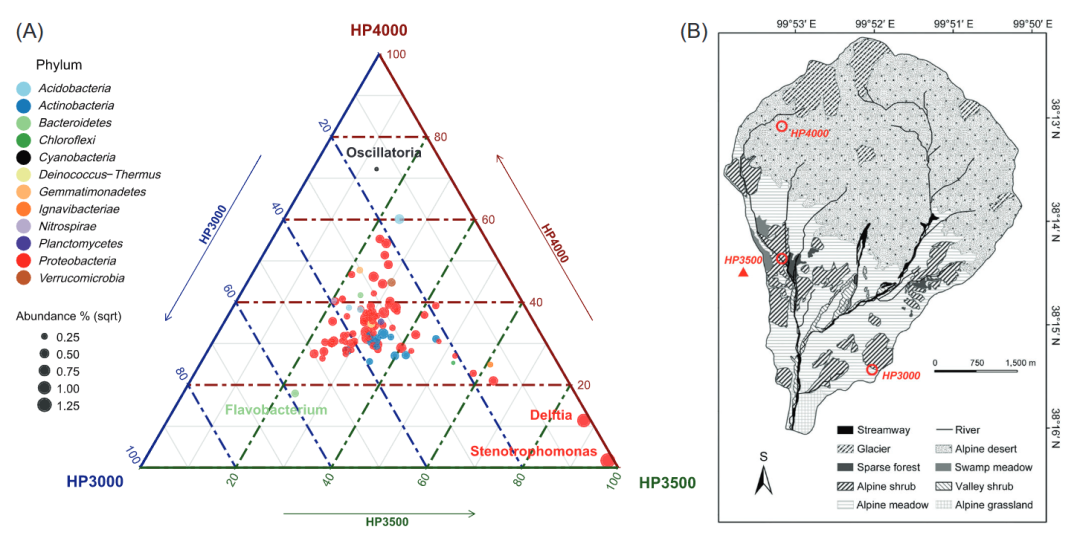
图2. A.三个不同海拔冻土中优势属(>0.5 ‰)的不同相对丰度。B.研究区域及采样点
尽管核心群落具有保守性,但值得注意的是,一些属明显受到区域环境因素的富集:在HP3000样品中富集的Flavobacterium;在HP3500样品中富集的Stenotrophomonas和Delftia;在HP4000样品中富集的Oscillatoria。在HP4000样品中富集的Oscillatoria属蓝藻门(Cyanobacteria),其可通过光合作用获取能量,能够在太阳辐射强的高海拔、低纬度地区生存。功能分析进一步证实,HP4000样品中蓝藻的固碳基因相对丰富度较高(图S8)。除卡尔文循环外,其他一些碳固定途径的基因,如还原柠檬酸循环(Arnon-Buchanan循环)、二羧酸-羟基丁酸循环和3-羟基丙酸双循环也在高海拔冻土中被检测到具有较高的相对丰富度。这些检测到的CO2固定的多种途径可能意味着冻土中蓝藻涉及到了多种利用光的途径。在高太阳辐射地区,蓝藻是主要的光利用生物,并供养了大量多样的异养微生物。
此外,在HP3500样品中富集的Stenotrophomonas和Delftia,分别占群落的4.95%和2.96%,该采样点的样品中硝态氮的浓度高达320mg/kg(表S1)。以往的研究表明,Stenotrophomonas在氮循环中具有重要的生态作用,它可以通过同化作用利用NH4+、NO2-和NO3-,但是不能将它们转化为N2。此外,当N2为唯一氮源时,它能固定氮气。冻土融化期中的微生物群的RNA-seq数据也揭示了它在硝酸盐氨化过程中的积极作用(图3),和氮含量对冻土群落的影响。根据之前的研究,Delftia可以将硝酸盐还原为亚硝酸盐。Delftia的硝酸盐还原基因参与反硝化、异化硝酸盐还原成氨(DNRA)和完全硝化过程(图S8)。Delftia的这种硝酸盐还原能力与某些有机化合物的降解有关。值得注意的是,这两个最主要的硝酸盐代谢属都不能进行完整的反硝化作用,这就是说在冻土中,硝酸盐往往直接转化为氨,而不是以N2的形式释放到大气中,可能会反过来促进土壤中氮的积累。
冷冻和融化冻土中微生物活性的转变
在非组装的ONT long-read宏基因组方法的帮助下,我们能够在前所未有的程度上解释冻土微生物群落的代谢潜力。论文补充材料中显示了三个海拔高度冻土中完整的微生物群落功能轮廓(图S9)。本研究重点研究了HP3500冻融冻土中的氮和甲烷代谢的转录活性,这两类功能在生物地球化学循环中发挥着重要作用。
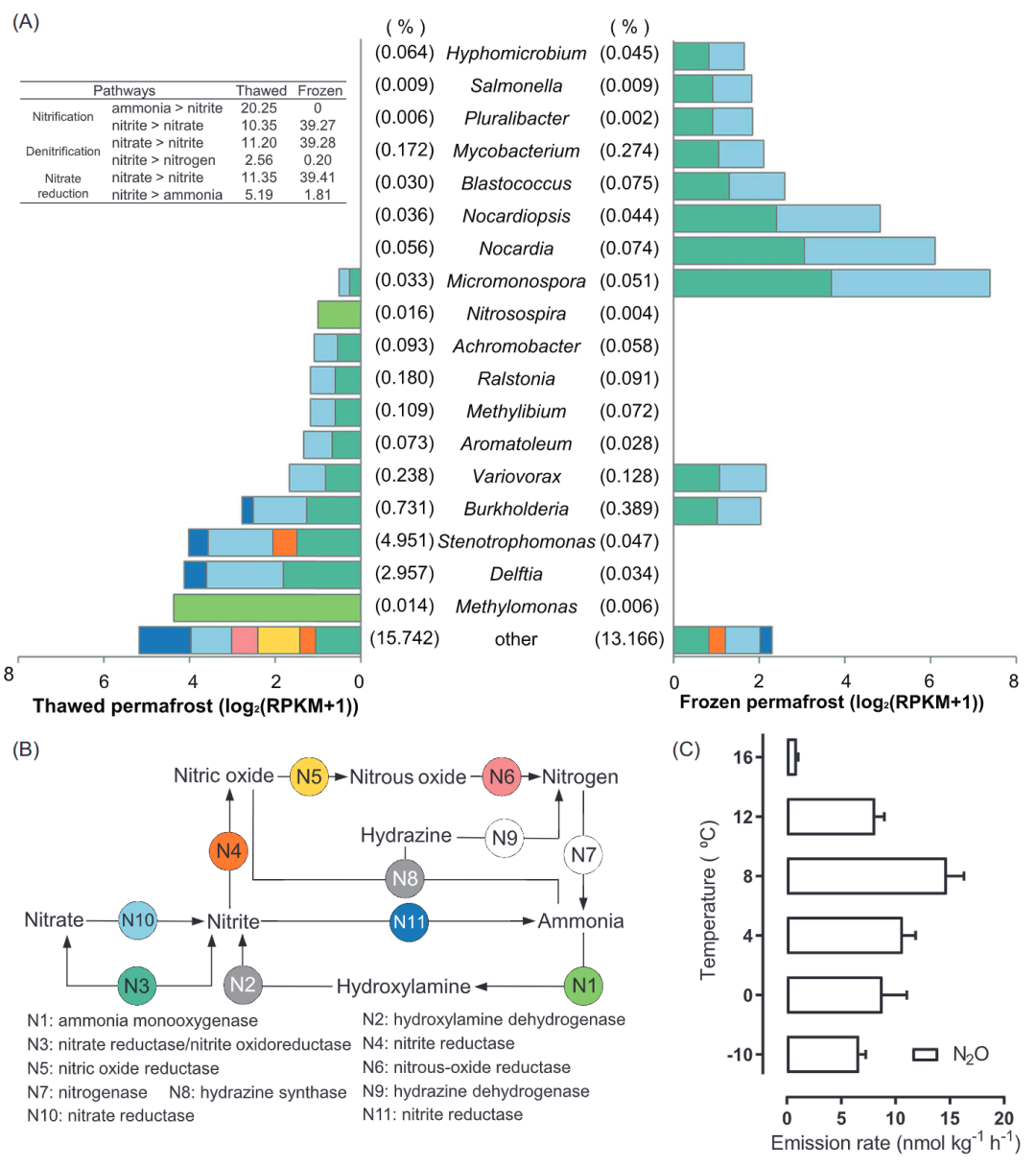
图3. HP3500采样点融化和冷冻冻土中氮代谢的转录活性。
A.参与氮代谢的不同微生物的转录活性。B.氮代谢主要通路和关键基因。C.冻土培养实验中不同温度下的N2O排放速率
氮循环
图3显示了HP3500冻土中氮的主要代谢途径的转录活性(RPKM)。总体而言,冻土融化期土壤中存在完整的硝化、反硝化和DNRA途径,而冷冻期冻土中不存在完整的反硝化途径。冷冻期和融化期均不存在完整的固氮过程,而两者均存在部分厌氧氨氧化过程。之前的研究表明,由于冻土环境中没有新的能源补充,简单而不稳定的可用性基质会随着时间的推移逐渐降低,这种寒冷的、能源有限的环境使得碎屑生物质被微生物作为主要碳/氮源进行循环利用。毫不意外的,本研究未检测到冻土中微生物的固氮过程,以及DNRA通路在冻融冻土中均表现出最高的转录活性,尤其是涉及从硝酸盐到亚硝酸盐转化的基因(图3中的N3和N10)。与反硝化作用不同,DNRA可保存系统中生物可利用的氮,产生可溶的铵而非不可利用的氮气。DNRA过程代表了一种防止微生物在土壤系统中消耗氮的方法,DNRA过程可以与电子供体(如有机物、氢、硫化物、甲烷和铁)氧化结合,当电子供体相对于硝酸盐过量时,DNRA似乎比反硝化更受青睐。我们观测到的强烈的DNRA途径表明,冻土的氮代谢趋势是一个封闭循环,而不是其他土壤微生物群落,如施肥土壤和河流沉积物那样释放氮气到大气中。冻土冷冻期的DNRA转录活性(RPKM为41.22)高于融化期(RPKM为16.54),这表明在冷冻状态下微生物仍具有较强的活性。以前的研究也显示,在寒冷的栖息地如冰川和永久冻土,仍然有活跃的微生物。
尽管在高海拔冻土中,整个氮循环是趋于封闭的,但在冻土融化期的表层土壤中,微生物的反硝化活动增加(图3)。由于反硝化是一种典型的异养过程,冻土融化期中碳可利用性的增加有助于其在冻土中的活性。冻土融化期(约15℃)的气相测量显示,N2O这种强大的温室气体,表现出一个较慢的消耗速率(18.8 nmol m-2 h-1,图S10),其产生可能是由于反硝化活动的增加。同时在冻土融化期观察到明显的N2O还原的转录活性,而在冻结期中表现不明显(图3)。结果表明,冻土融化期温度为15℃时,N2O更容易被微生物消耗,更倾向于转化为N2。我们的冻土培养实验进一步证明了这一现象,冻土培养温度从−10°C到8°C, N2O排放量增加,温度继续上升到10°C以上(8°C到16°C),N2O排放量减少(图3C)。这主要是因为低温环境对N2O消耗酶(N2O还原酶)的抑制程度大于对N2O产生酶(NO3-、NO2-、NO还原酶)的抑制程度,且在10℃以上N2O还原酶活性的恢复导致培育实验中观察到的N2O排放减少.
此外,参与氮代谢的微生物群落在融化和冷冻状态之间有明显的变化。对北极/南极冻土的研究表明,冷冻是影响冻土微生物群落和功能的关键因素,冻土活动层的解冻会引起包括C、N代谢基因在内的微生物功能基因的快速变化。融化过程中,寡营养微生物活性降低,共营养微生物活性增加,表明融化过程中被冻结的有机质变得易利用,为微生物提供了能量来源。这些结果可以解释我们观察到的现象,即在冻土融化期,反硝化代谢的转录更活跃(图3),因为大多数反硝化菌是异养的。因此,在冻土冷冻期参与DNRA通路的微生物从Proteobacteria的共养/异养菌群(如Delftia, Stenotrophomonas和 Burkholderia)转变为寡营养养/自养菌群(如Micromonospora、Nocardia和Nocardiopsis)。
亚硝化螺旋菌(Nitrosospira)是冻土融化期群落中的活性氨氧化细菌(AOB),而冻土冷冻期中未发现活性AOB,这表明冷冻期中活性AOB受到明显的抑制。此外,我们在冻土中没有发现活性氨氧化古菌(AOA)。与之前研究类似,在寒冷环境如西伯利亚东北部的高海拔湖泊和冻土中发现了AOB主导的氨氧化。Nitrosospira是一种典型的生活在多种环境中的AOB,虽然生活污水处理厂中的典型Nitrosospira更喜欢温暖的生长环境(25-30℃),近年来的研究发现某些Nitrosospira在寒冷环境中也具有适应性和富集性,我们的结果也确认了这一发现。
此外,甲基单胞菌(Methylomonas)也是冻土中可能存在的氨氧化细菌,Methylomonas是一种需氧甲烷嗜氧菌,从甲烷中获取碳和能量,冻土在解冻过程中,有机质分解生成甲烷,可能有利于Methylomonas的富集。在之前的研究中发现,甲基单胞菌中有一种编码序列分化的颗粒甲烷单加氧酶,也可能参与氨氧化。
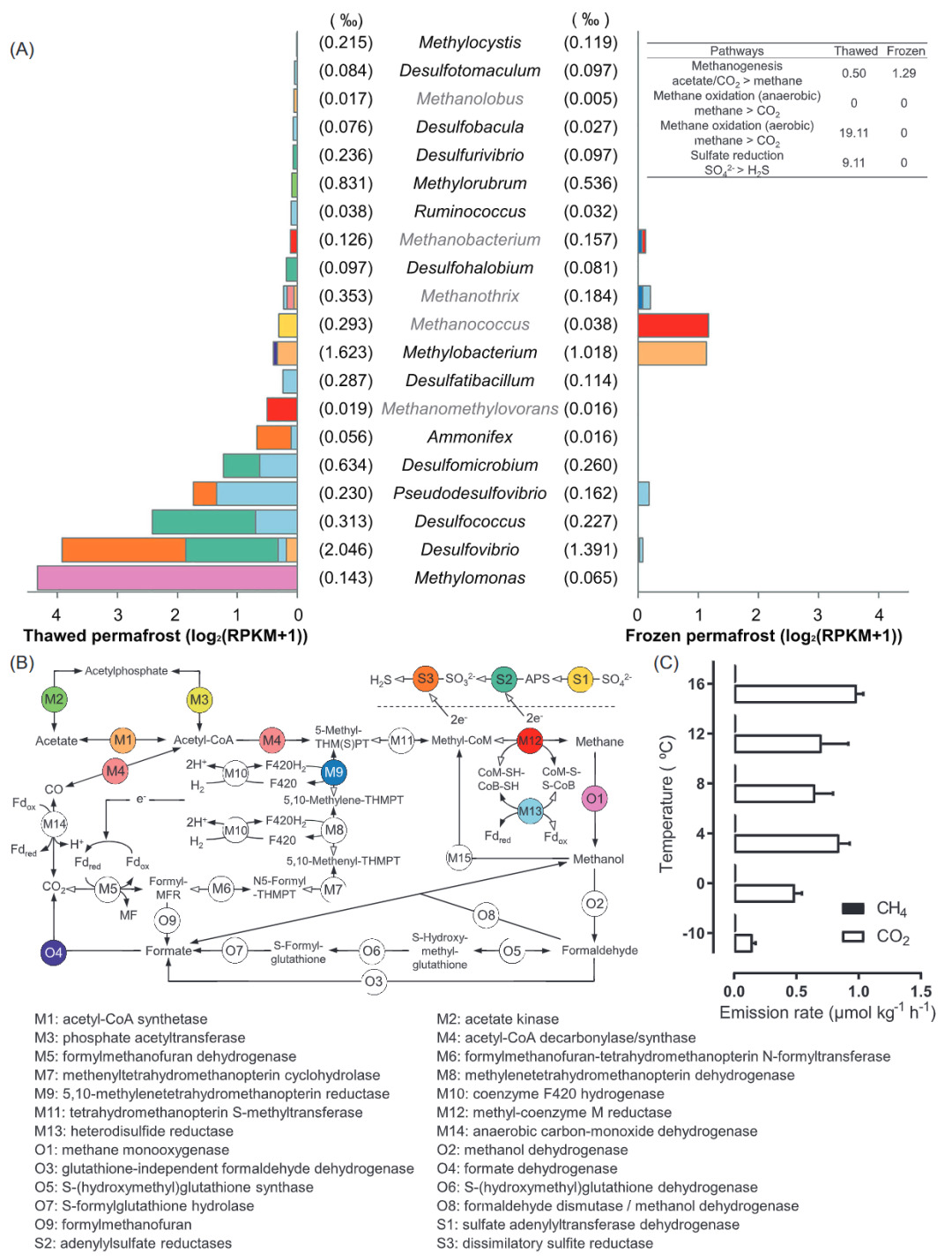
图4. HP3500采样点融化和冷冻冻土中甲烷代谢的转录活性
A.参与甲烷代谢的不同微生物的转录活性。B.甲烷代谢主要通路和关键基因。C.冻土培养实验中不同温度下的甲烷和二氧化碳的排放速率
甲烷代谢
因为甲烷代谢中的一些酶可以催化多种途径的反应,根据之前的研究,我们采用人工过滤的方法对这些特定的甲烷代谢微生物进行了筛选。如图4所示,没有发现完整的产甲烷和产甲代谢的途径,这可能是由于冻土产甲烷菌和产甲烷营养群落丰度较低(融化期约为0.8‰,冷冻期约0.4‰)所致。然而,甲基辅酶M还原酶(Mcr)作为产甲烷古菌中催化产甲烷反应的限速及最后一步的关键基因,其在几个产甲烷菌属中都有大量转录活性,这表明冻土中的产甲烷活动比较活跃。
冷冻和融化冻土中的产甲烷群落和途径存在明显差异。融化冻土中主要的活性产甲烷菌是Methanomethylovorans(Methanosarcinales目,同时具有乙酸和氢营养型产甲烷作用),而冷冻冻土中是甲烷球菌Methanococcus(Methanococcales目,严格的氢营养产甲烷菌)(图4)。此外,在融化冻土中检测到的产甲烷途径在Methanothrix(M1, M4和M13)和Methanolobus(M1)中,其都隶属于Methanosarcinales目(图4B),具有乙酸型产甲烷途径。然而,在冷冻冻土中,严格的氢营养型产甲烷菌(Methanococcus和Methanobacterium)更常见,以及Methanosarcinales的产甲烷途径也从乙酸型转变为氢营养型(Methanothrix的M9,M13,图4A右侧)。我们的结果进一步证实了之前的猜测,即冻土融化将促进氢营养型转变为乙酸型产甲烷途径。在祁连山冻土中,微生物活动可利用的有机质浓度较高,多糖降解促进了产甲烷的活性,这可以解释祁连山冻土融化过程中产甲烷途径从氢营养型向乙酸型产的现象。
好氧甲烷氧化(AMO)是祁连山地区主要的methanotrophy过程,活性AMO仅在融化冻土中检测到(图4)。AMO活性主要来自Methylomonas(图4)。最近的研究表明,微氧环境可以为Methylomonas的生长提供足够的氧气。Methylomonas编码一种序列分化的颗粒甲烷单加氧酶,也可参与氨氧化。冻土中可参与到氨氧化和AMO中的活跃的Methylomonas,意味着冻土中的微生物群落在碳氮循环中的协同作用。除AMO外,厌氧甲烷氧化(AnMO)还可以通过逆氢营养型产甲烷途径将甲烷氧化为CO2。这些厌氧甲烷营养体通常属于Methanosarcinales (ANME-2和ANME-3)和“Ca.Methanophagales”(ANME-1)。AnMO反应需要与能通过还原硫酸盐、硝酸盐或铁提供电子的共养细菌偶联。然而,在本研究中,虽然硝酸盐(图3)和硫酸盐(图4)的还原活性很高,但未检测到厌氧古菌甲烷营养菌。这种AnOM过程缺失的情况与之前对北极冻土的研究一致。
冻土冷冻期的甲烷产量与消耗比为1:38(基于关键基因的RPKM),表明微生物AMO能够有效地将甲烷转化为CO2,并减缓冻土融化甲烷的释放。这一结果与现场观察到的结果一致,即甲烷的排放速率为2.0 μmol m-2 h-1,而CO2的排放速率要高50倍(104.5μmol m-2 h-1,图S10)。此外,在不同温度下冻土的培养实验也证明冻土融化过程中会释放更多的CO2(图4C)。由于甲烷的温室效应比CO2强得多,Methylomonas对甲烷的生物转化可以缓解冻土融化引起的温室气体排放。
结 论
为了解决ONT宏基因组数据组装导致的大量数据丢失问题,我们开发了一个ONT long-read校正和注释流程(FUNpore)。这种基于校正的注释策略在功能注释中具有良好的准确率和回收率,并且可以提供更多的冻土微生物信息。对高海拔冻土进行的MinION宏基因组测序表明,虽然在不同海拔高度的冻土中有78.4%的微生物属是共有的,但硝酸盐浓度和太阳辐射可导致一定程度的群落分化。冻土中的氮代谢具有封闭循环的趋势,尤其是冻土的冷冻期。冻土融化过程中反硝化活性增加,但融化过程中产生的N2O更有可能转化为N2。冻土融化过程中产甲烷途径由H2营养型转变为乙酸型,且Methylomonas的好氧氧化作用可显著减少甲烷的排放。综上所述,在全球变暖的背景下,融化冻土的增加将显著改变冻土中氮和甲烷的代谢,然而微生物可以作为生物过滤器来缓解由于冻土融化引起的温室气体排放。
局限性和注意事项
在本研究中,我们基于ONT long-read的宏基因组和Illumina的RNA-seq宏转录组,揭示了微生物对高海拔冻土冻融循环的响应。虽然我们基于校正的注释策略在微生物活性鉴定中非常有效,但是一些限制可能会影响我们的结果。首先,我们的样品来自一年内采集的冻土样品,研究冻融循环还需要进行长期的研究才能更好的支撑当前的结果。第二,我们的注释策略基于比对的(BLASTP)的条件下准确率非常高,而基于隐马尔可夫模型(HMM)的工具就会有很多错误注释。因此,目前流行的基于HMM的工具如GroupM、HMMer、Pfamscan等不能用于预测校正后的ONT long-read的功能。此外,由于ONT long-read实际上比组装的contigs长得多,基于校正的策略可以将微生物的功能注释到属水平(属水平上主要代谢能力通常是保守的)。然而,后续仍需要对校正后的ONT long-read进行进一步的基因组分箱分析(binning),以研究微生物的一些不保守功能,如致病性和抗生素耐药性等。
研究方法
研究区域及冻土样品采集
采样地点是青藏高原东北部的祁连山(N38º13'-38º16',E99º50'-99º53'),海拔约4800米。该地年平均气温在−5~11℃,7月的最高日平均气温约为23℃,1月的最低日平均气温约为−20℃。常年的低温使山上的积雪形成了广阔的冰川。我们在不同海拔(3000、3500和4000米)的冻土活土层选取采样点,并分别命名为HP3000、HP3500和HP4000。我们在2018年8月开展了3个取样点的融化冻土采样活动,并于2018年11月在HP3500处采集冷冻冻土。每个土壤样品在季节性冻融过程而形成的冻融坑表面采集,并且是在三个或多个地点采集平行样品。将样品混合均匀后密封在50 mL灭菌聚丙烯管中,把每份土壤样品分为三部分:一部分用于现场提取DNA,即立即用于Oxford Nanopore测序;第二部分(约5 g)立即用50 mL RNA-later(Invitrogen,USA)浸泡固定,带回实验室提取RNA;最后一部分储存在冰中,再运输到实验室进行后续分析。关于永久冻土样品的理化性质分析方法在表S1中作了论述。
DNA提取和野外MinION测序
每个样品都使用DNeasy PowerSoil Kit(QIAGEN,德国)提取三个生物重复的DNA。DNA浓度由Qubit 2.0仪器(Thermo Fisher Scientific, Wilmington, DE, USA)测定,用AMPure XP beads(Beckman Coulter)从每一个重复样品中提取等量的DNA并对其纯化。
从每个样品中取约2.0 μg纯化的DNA,使用SQK-LSK108 1D连接基因组DNA试剂盒进行测序文库准备,并在流程上稍作修改,详细过程请参考论文。最终共使用5个测序芯片对5个样本进行独立的MinION测序,其中包括一个生物重复样品。平均每个样品获得了5.5 Gb clean reads数据(fasta格式)(表S2)。
现场气体样品采集
为了测定土壤-空气界面N2O、CO2和CH4的通量,我们采用改进的静态室法测定了气体交换速率,在土壤上放置两个装有排气孔的腔室,以20分钟间隔收集气体。详细过程请参考论文。
实验室提取RNA
冻土样品在RNA-later中用冰运到实验室。使用RNeasy PowerSoil总RNA试剂盒(QIAGEN,德国)提取总RNA。详细过程请参考论文。
Illumina测序
DNA测序详细过程请参考论文。平均每个样品得到44.3 Gb的clean reads(fasta格式)。RNA测序详细过程请参考论文。平均每个样品得到38.5 Gb的 mRNA reads(fasta格式)。所有原始测序数据均上传至EBI(ENA)数据库,登录号为PRJEB38431。
ONT reads校正
校正的详细过程请参考论文。此外,使用两个数据集校正后的ONT reads来评估基于KEGG Orthology(KO)的功能预测的准确率和回收率。第一个数据集是20株分离菌株的ONT和Illumina全基因组序列。菌株的混合组装作为评估的黄金标准(表S3)。第二组数据集为从NCBI基因组数据库下载的315个全基因组(表S4)。为保证评价结果的代表性,每个微生物门的基因组数相同。我们在这些下载的基因组中随机引入5%的误差来当作模拟的ONT数据集,然后将修正后的ONT reads的功能预测与下载的基因组进行比较以进行评估。
测序数据分析
所有的分类和注释的步骤都在我们的FUNpore流程中(https://github.com/sustc-xylab/FUNpore.git)(图1A)。详细过程请参考论文。
冻土培养实验
取200g冻土置于带橡胶塞的血清瓶中,在-10℃、0℃、4℃、8℃、12℃、16 ℃温度梯度下连续培养,每个温度恒温24 h,并在培养开始和结束时采集气体样品。每次采样后都用N2冲洗瓶子,以平衡在气体采样时产生的负压。培养实验设置了两个平行。
气体检测与分析
采用配备了热导检测器(TCD)的气相色谱仪来检测N2O、CO2和CH4。色谱柱为HayeSep-Q,载气为高纯氦(99.999%),流速为30mL min-1。烘箱和TCD的操作温度分别为120℃和200℃。
气体的排放或消耗率由下式计算
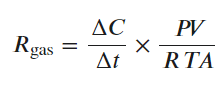
式中△C/△t表示气室中某种气体累积率的斜率(ppm/h),斜率为正表示土壤释放气体,斜率为负表示土壤消耗气体;P表示大气压力(atm);V表示气室容积(L);R表示气体常数(0.0821 L atm K-1 mol-1);T表示气体温度(K);其中A表示箱体在土壤表面的面积(m2)或培养的永久冻土的重量(kg)
数据可用性声明
所有测序数据已上传至EBI,提交编号为PRJEB38431。FUNpore脚本已上传至GitHub(https://github.com/sustc-xylab/FUNpore)。补充材料(图片摘要、幻灯片、视频、中文翻译版本和更新材料)可通过DOI或iMeta网站http://www.imeta.science/获取。

党晨原(第一作者)
● 北京大学博士、华中科技大学讲师、iMeta期刊青年编委
●多年来从事环境微生物、环境基因组、环境生物技术等相关领域的研究。主要使用分子生物学、生物信息学等方法研究自然和人工系统环境中微生物驱动的物质和能量循环的机理和生态效应,以及其与环境中新型污染物(抗生素、抗性基因等)的相互作用规律。以第一或通讯作者在Environmental Science & Technology、Water Research等环境领域顶级期刊发表研究论文20余篇,撰写科学专著章节(Invited Book Chapter)1篇

夏雨(通讯作者)
● 香港大学博士、南方科技大学助理教授(副研究员)、iMeta期刊青年编委
● 研究方向集中于:利用新一代测序技术与生物信息学大数据分析相结合的手段研究环境微生物群落组成和协同作用关系对(1)生物处理技术运行效率; (2) 致病菌的环境分布以及抗生素抗性基因在环境中的迁移转化; (3)环境物质循环的驱动机理和生态效应的影响。已在The ISME Journal、Environmental Microbiology、Biotechnology for Biofuels、Environmental Science & Technology、Water Research等重要期刊发表SCI论文20余篇。曾担任美国微生物协会香港地区青年大使以及香港大学研究生协会常务秘书长
(▼ 点击跳转)
iMeta文章中文翻译+视频解读
iMeta封面 | 宏蛋白质组学分析一站式工具集iMetaLab Suite(加拿大渥太华大学Figeys组)
▸▸▸▸
iMeta | 深圳先进院马迎飞组开发基于神经网络分析肠道菌群的方法
▸▸▸▸
iMeta | 南医大陈连民等综述从基因组功能角度揭示肠菌对复杂疾病的潜在影响
▸▸▸▸
iMeta | 北大陈峰组综述口腔微生物组的标准化研究:从技术驱动到假说驱动
▸▸▸▸
iMeta | 电子科大林昊组开发蛋白质赖氨酸乳酸化位点预测工具DeepKla
▸▸▸▸
iMeta | 南昌大学丁霞等-水产养殖系统对中华鳖微生物组和肠道代谢组的影响
▸▸▸▸
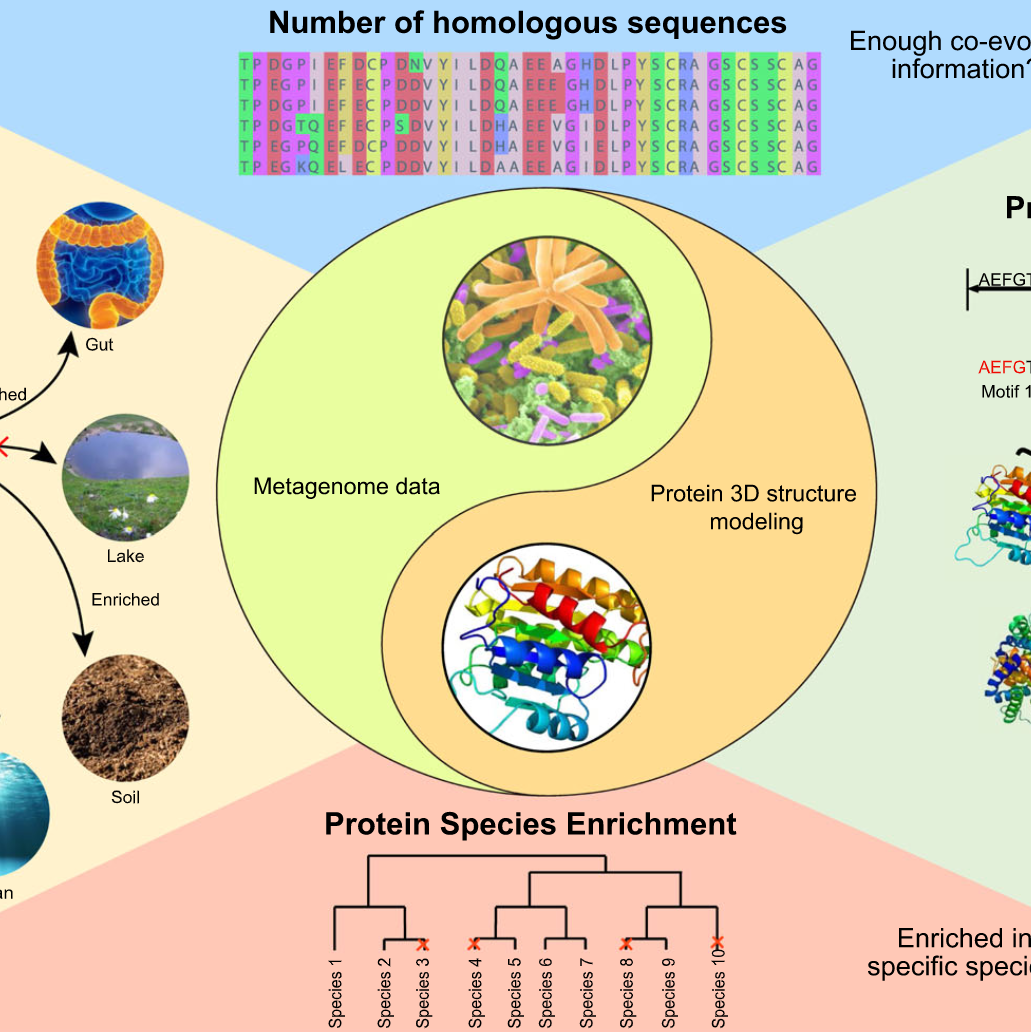
iMeta | 华中科大宁康组综述用于蛋白质结构预测的宏基因组定量分析
▸▸▸▸
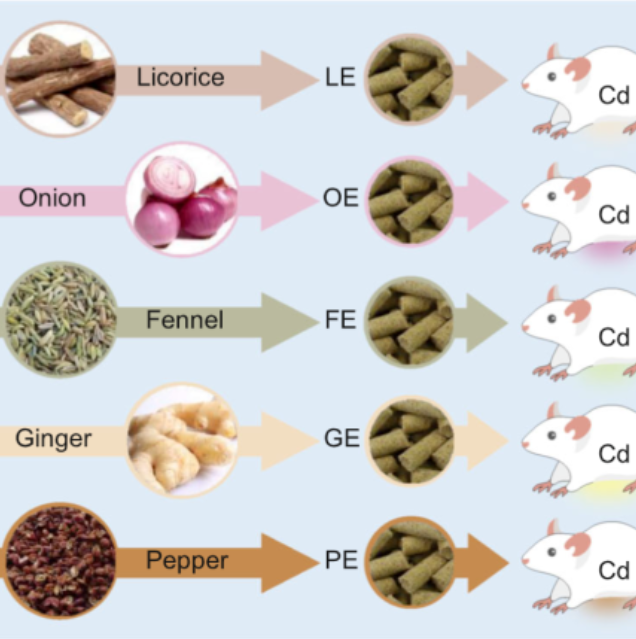
iMeta | 中科院李小方等膳食甘草促进小鼠镉解毒并调节肠道菌群代谢
▸▸▸▸
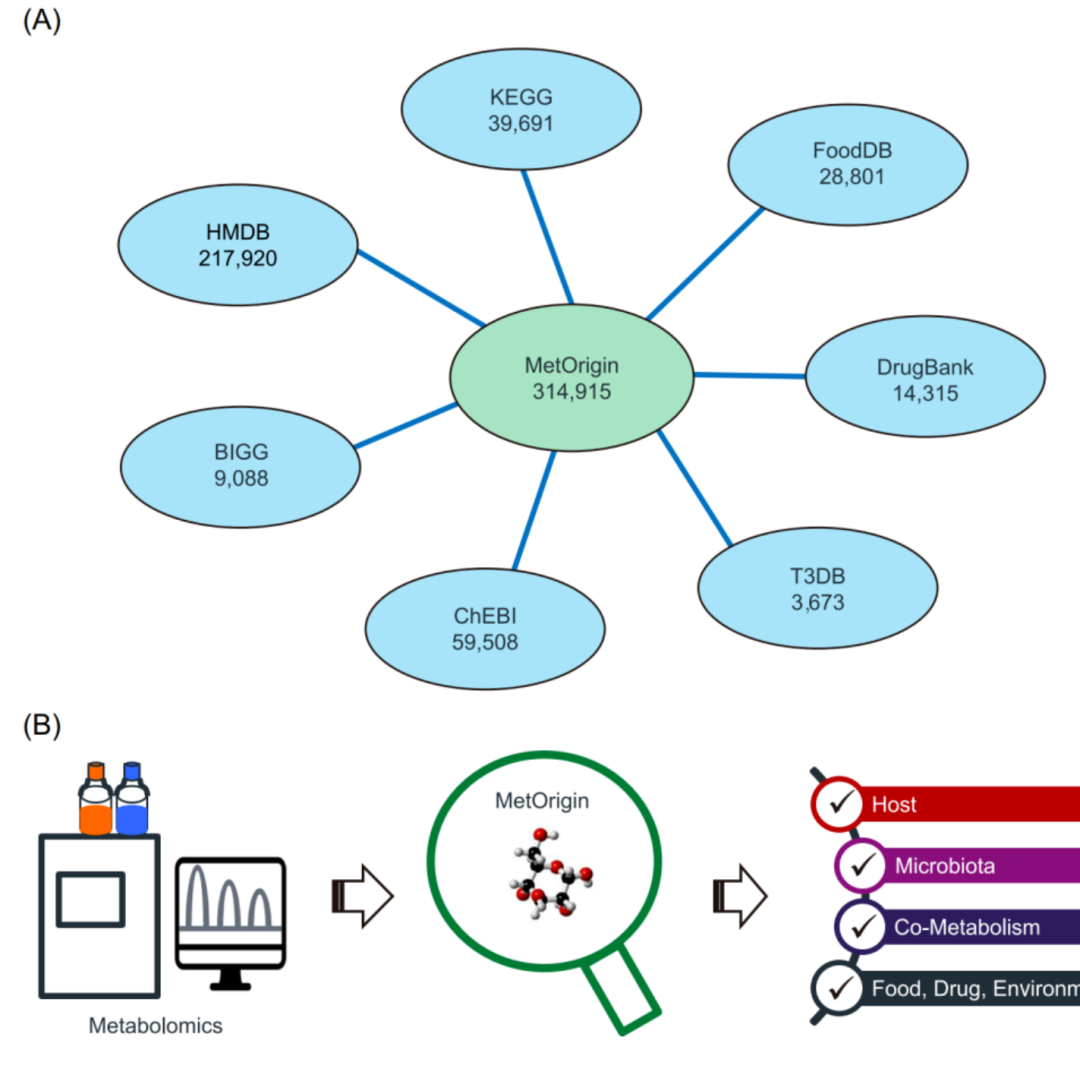
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析
▸▸▸▸
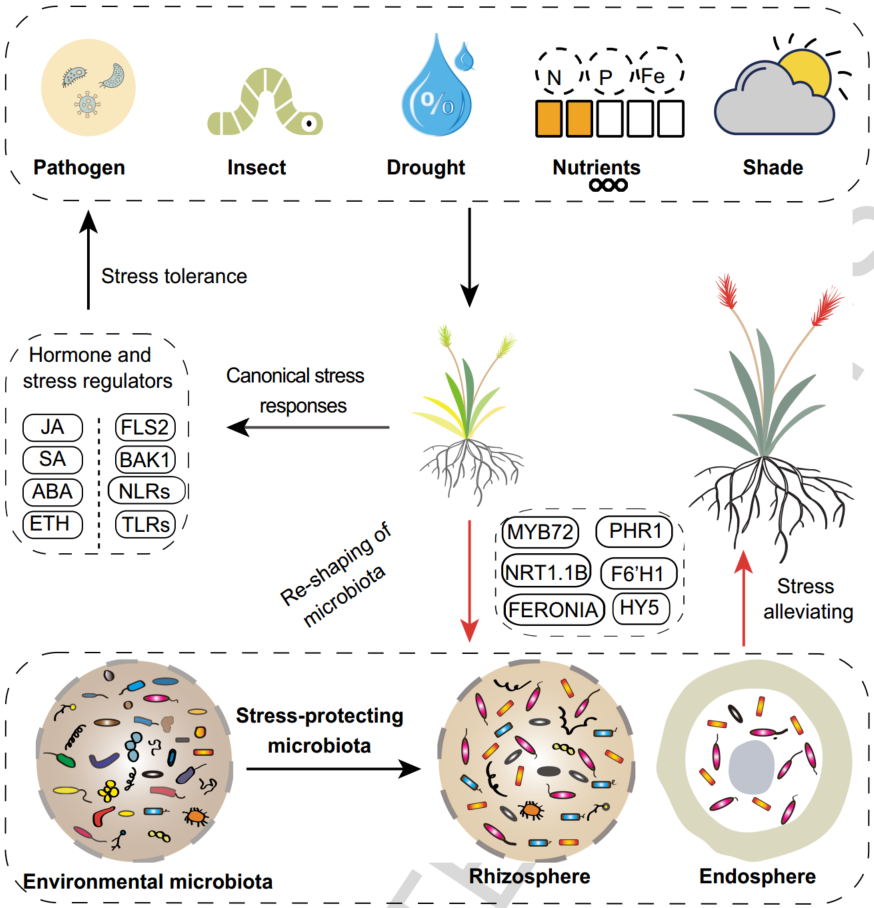
iMeta | 南科大宋毅组综述逆境胁迫下植物向微生物组求救的遗传基础(附招聘)
▸▸▸▸
iMeta:高颜值高被引绘图网站imageGP
iMeta教你绘图
使用ImageGP绘图热图Heatmap
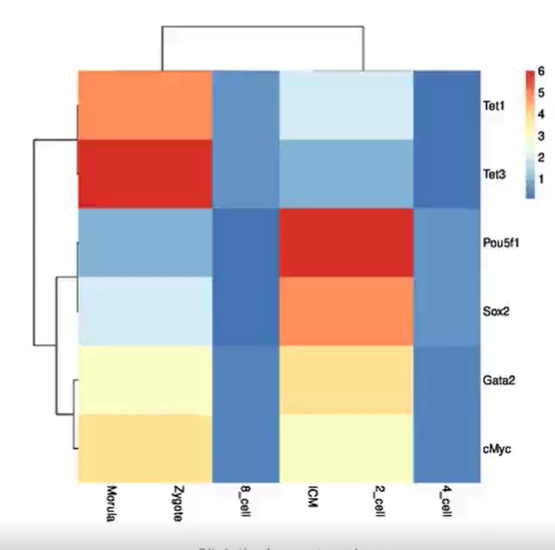
▸▸▸▸
使用ImageGP绘图富集分析泡泡图
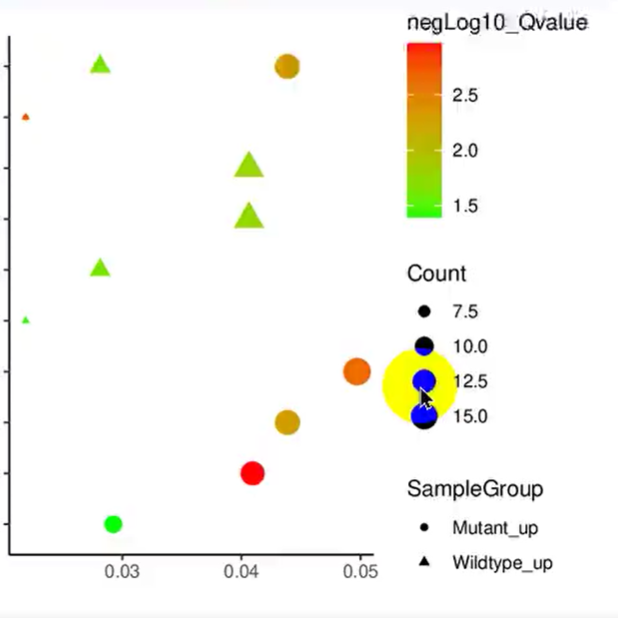

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science
微信公众号
iMeta
责任编辑
微微