点击上方“视学算法”,选择加"星标"或“置顶”
重磅干货,第一时间送达
本文来源:“TOP大学来了”综合自“张国捷教授个人网站、iNature、浙江大学”
2月23日,据浙江大学,丹麦哥本哈根大学终身正教授张国捷已于本周一全职加盟浙江大学,任生物演化研究中心讲席教授。
据悉,张国捷教授近期科研成果相当丰硕,2020年11月到2021年4月期间,张国捷教授团队在不到半年的时间内,连续发表了6篇Nature/Cell!其中3篇封面文章!2021年4月发表的Nature论文被选为杂志封面,2021年2月背靠背发表两篇cell,2020年11月,背靠背发表两篇Nature,并被选为杂志封面!据不完全统计,截止目前,张国捷教授发表的Nature, Science, Cell论文,已经超过30篇!如此高质高产,堪称传奇!
张国捷教授在其个人网站标注已加盟浙江大学
张国捷教授简介
所属学科生物学
张国捷,浙江大学生物演化研究中心讲席教授。哥本哈根大学、中国科学院昆明动物研究所兼职教授。2004年毕业于厦门大学,获学士学位。2010年毕业于中国科学院昆明动物研究所,获博士学位,2012年起在丹麦哥本哈根大学担任助理教授,2017年转为终身正教授。
迄今在国际知名杂志Nature, Science, Cell等期刊共发表研究论文200余篇,其中有数十篇为通讯作者身份发表在Nature, Science, Cell。论文总引用数超过3万次,自2018年连续多年入选全球高被引学者。实验室具有自由的学术研究气氛,支持课题组成员独立探索感兴趣的课题。课题组组织了多个国际科研协作项目,与国内外世界一流的研究单位和学者长期开展合作,为学生提供国际化的发展空间。
半年不到连发6篇Nature/Cell!
其中3篇封面!
由张国捷带领深圳华大生命科学研究院的生物多样性基因组学团队于2020年11月至今,共发表4篇Nature和2篇Cell论文,其中3篇为杂志封面。
该团队致力于基因组学以及生物多样性和社会行为演化等相关领域,发起并参与了多项研究项目:万种鸟类基因组计划、全球蚂蚁基因组联盟计划、万种脊椎动物基因组计划、地球生物基因组计划。2021年4月28日,Nature在线发布了张国捷团队的论文并选为杂志封面,标题为 “Evolutionary and biomedical insights from a marmoset diploid genome assembly” 。研究利用家系基因组测序(trio-binning approach)数据,组装出普通狨猴(Callithrix jacchus)的父母本两套高质量基因组。因狨猴与人类的诸多共同特征,狨猴是人类疾病研究模型之一,可应用到神经学、生殖动物学、干细胞研究、感染性疾病等多方面研究中。该文章利用狨猴高质量基因组,对比了亲本遗传基因组,分析了胚系变异;并发现影响狨猴独特生物学特性的相关基因,证明家系基因组测序法的实用性——可大力推进人类疾病学研究。
正常哺乳动物的染色体是成对存在,分别来自父母双方。这两条同源染色体存在遗传差异。但在DNA测序时,由于算法存在一定的差异性,所以只保留其中一种样本。因此传统染色体测序法无法组装出双倍基因组,只能给出随机混合父或母本遗传信息的单倍基因组。该研究采用家系基因组测序法,与传统测序法不同,该方法将染色体中父母本两套基因组独立组装,提供高染色体测序的质量。
家系基因组测序法可直接比较两个亲本遗传的基因组,利于辨识父母本套基因组的基因特异,包括单碱基变异(SNV)、基因的插入与缺失(INDELs)、大型基因组结构性变异 (Large SVs) 。基因组结构性变异贡献了大量遗传多样化,具有重要的进化学和医学意义。研究证实家系基因组测序法可为父母本套等位基因差异提供高准确性。从实验对象完整基因组中发现3.47百万个单碱基变异以及大约232,000 短基因插入与缺失。其中96.5%的单碱基变异与短序列匹配算法(short read mapping)结果相符。通过随机选择,99.6%的单碱基变异和95.2%的基因插入与缺失与PCR实验相符。研究通过比较两个单倍体基因组,综合共116,631 结构性变异(SVs>50 bp),发现被实验对象的体细胞杂合率大约为1.36%。胚系变异是基因多样性的又一导致基因多样化的因素,并有推动生物进化和多种基因疾病的作用。然而因传统基因组装法无法区分父母本套基因组,导致部分可追踪到亲本起源的变异无法被研究。该研究使用的完整二倍体基因组装法,可借鉴父母单倍体基因组,侦测从头胚系突变(de novo mutation)。研究从父系和母系基因组的约41%的可调用位点中检测到9个有效的从头胚系变异。狨猴的从头胚系突变的父系与母系比例为2:1。该比例低于人类的4:1,却和与人类为近亲的夜猴的比例近似(2.1:1)。研究发现狨猴每代每个基因的从头胚系变异率为0.43*10^-8。研究还通过完整二倍体基因组装法解析了狨猴的X和Y染色体。通过比较人类与猿猴的性染色体,发现狨猴的新性别分化区域(SDR)膨胀是一个进化上的年轻事件。还观察到狨猴的性染色体的假常染色体区域(PAR)上GC-碱基含量相比人类更高。狨猴假常染色体(PAR)的杂合率(0.52%)为常染色体的平均杂合率(0.12%)的4.3倍,这表明较短的狨猴PAR中更强烈的重组导致更多突变。通过比较猿猴和人类的X染色体上的扩增锥基因(AGs)发现,在灵长类进化过程中,性关联的扩增锥基因经历了非常动态的复制过程。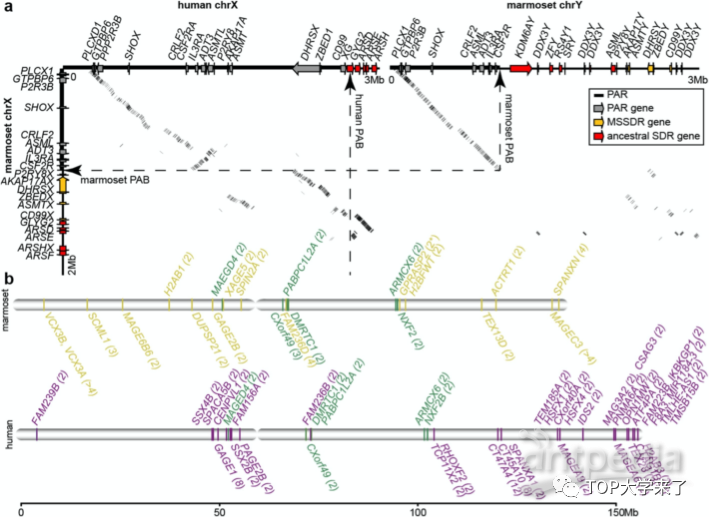
灵长类的Y染色体结构变化较为快速。研究中比较了人类与狨猴的Y染色体的雄性特异性区域(MSY)并且发现22个人类MSY基因子狨猴基因中缺失。研究人员推测这些狨猴缺失的基因可能降低精子间竞争,并与猿猴一夫一妻制社会有关。文章指出虽然普遍认为狨猴的精子形成模式与人类相似,但可能实际存在一些关键差异。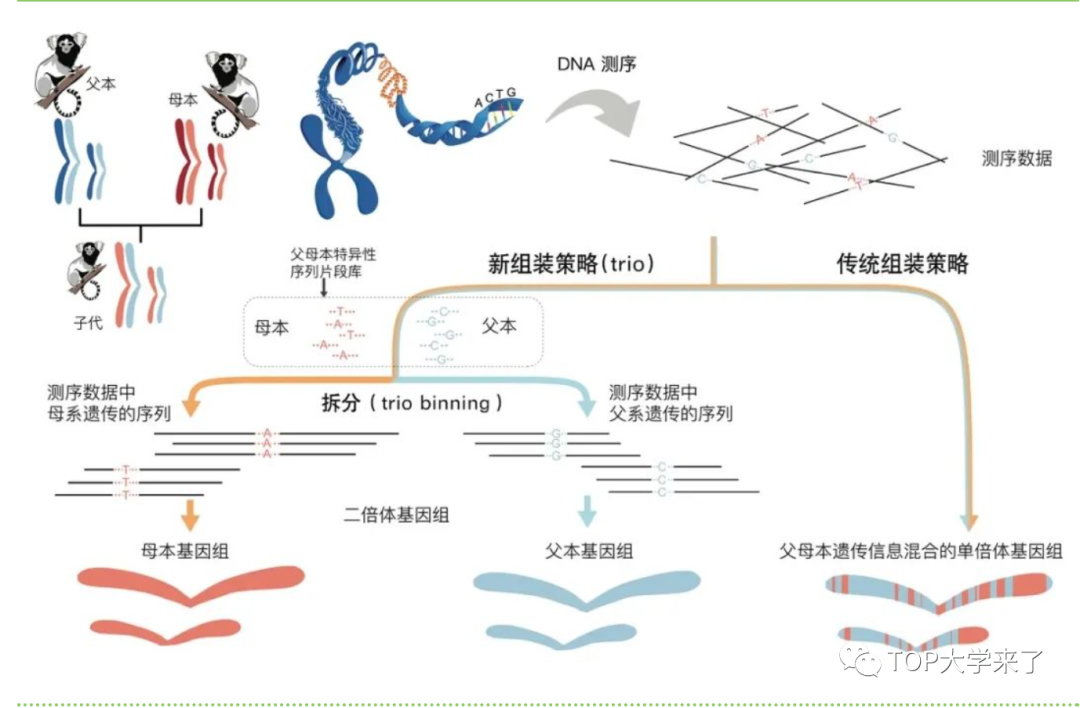
图3:子代个体的两套染色体一套来自于母亲,一套来自于父亲。传统组装策略获得的是遗传信息混合的基因组,新策略则可以通过父母本特异的序列得到遗传自父母本的两套完整的基因组数据(杨琛涛和周旸 绘)
总而言之,研究指出家系基因组测序法结合长阅读测序,产生的二倍体基因组具有高准确率。相比于大多数基因组研究方法——仅使用杂合SNV——所发现的杂合率,家系基因组测序法发现的杂合率高10倍。并且此法获得的二倍体组合包含更完整的两条性染色体序列。文章认为,这种策略可以更好地为杂合度高的二倍体物种生成高质量基因组。该研究是二倍体物种 “完美基因组” 的一个成功范例。2021年2月4日,张国捷团队在全球权威期刊Cell分别发表了题为“Tracing the genetic footprints of vertebrate landing in non-teleost ray-finned fishes”和 “African lungfish genome sheds light on the vertebrate water-to-land transition”的研究论文 。两篇论文分别通过研究原始辐鳍鱼类和非洲肺鱼的基因组,为脊椎动物从水生进化为陆生的进化之路填补了新的见解和关键资源。

化石证据表明,在总鳍鱼类和条鳍鱼类的祖先之前,许多与陆地进化相关的特征和功能就已存在。在前一篇论文中,研究分析了多鳍鱼、鲟鱼、弓鳍鱼、雀鳝等原始辐鳍鱼类的基因序列,证明颌类脊椎动物的祖先已经拥有支持空气呼吸的心肺系统的潜在基因网络。研究发现:1)通过与颌类脊椎动物基因组相比,原始辐鳍鱼类中存在四肢发育的增强子,这支持了相关调节四肢运动的遗传基础早在四足动物起源之前就已出现的假说。2)转录组分析证实了肺和鱼鳔之间的同源性,并揭示了原始辐鳍鱼肺相关功能基因的存在。原始辐鳍鱼具有检测空气分子的嗅觉受体,且拥有与血管新生通路相关的高表达基因。3)原始辐鳍鱼类的心脏系统具备人类心脏的动脉圆锥结构。研究发现在人类和多鳍鱼之间,心脏系统基因有保守的调控机制;并且首次找到调控Hand2基因的保守调控原件。肺鱼是四足动物现存的近亲,保留着水—地过渡的祖先特征。张国捷团队在非洲肺鱼一文中,进行了非洲肺鱼基因组的40gb染色体水平的组装,这是有史以来最大的基因组组装。肺鱼基因组的大尺寸主要是由于反转录转座子。超长基因的表达水平与其他基因相似,说明肺鱼已经进化出高转录效率以保持基因表达的平衡。研究通过研究呼吸系统中的肺表面活性剂蛋白相关基因的演化,发现:原始辐鳍鱼类已具备初步空气呼吸能力,后肉鳍鱼祖先呼吸能力增强,最终四足动物演化出成熟的呼吸能力。此外,研究确定了一个控制四足动物五指的潜在基因和调控元件——该原件在鱼、蛇、鸟和人类中皆有变异,还确定了肺鱼和四足动物共同拥有的负责抗焦虑的两个重要基因。2021年1月6日,张过捷团队在Nature发表了名为“Platypus and echidna genomes reveal mammalian biology and evolution”的研究论文。
团队利用单分子测序技术和多种物理谱图方法,获得了高质量的鸭嘴兽基因组。研究通过对比鸭嘴兽基因组和短鼻针鼹的基因序列,解析了单孔目动物的复杂性染色体系统的起源和多样性,这为哺乳动物的进化提供了关键见解。文章指出,单孔性染色体复合体起源于一种染色体环状配置,这种配置可能是通过多对古老的常染色体之间的非同源的片段交换形成。团队还认为,这种多对性染色体的形成,可能与多对性染色体之间明显高于常染色体的相互作用有关。文章最后,通过进一步的比较鸭嘴兽和针鼹基因组,揭示了与爬行类和哺乳类动物相比,单孔目动物在结合珠蛋白基因、泌乳基因和嗅觉、味觉基因上的显著差异。2020年11月11日,张国捷与加州大学圣克鲁斯分校的Benedict Paten为共同通信,在Nature以封面文章形式发表了两篇论文:“Progressive Cactus is a multiple-genome aligner for the thousand-genome era “和” Dense sampling of bird diversity increases power of comparative genomics “。

研究团队建立了适用于多物种的无参基因组对比和分析的新方法——Progressive Cactus。这一方法解决了多序列对比软件的弊端,即无法识别特定序列以及丢失同源区域。新算法极大提高了跨物种的对比效率,减少了序列的丢失。相较于之前的鸟类全基因组序列,由Cactus构建的363只鸟类全基因组对比序列在长度上提升149%。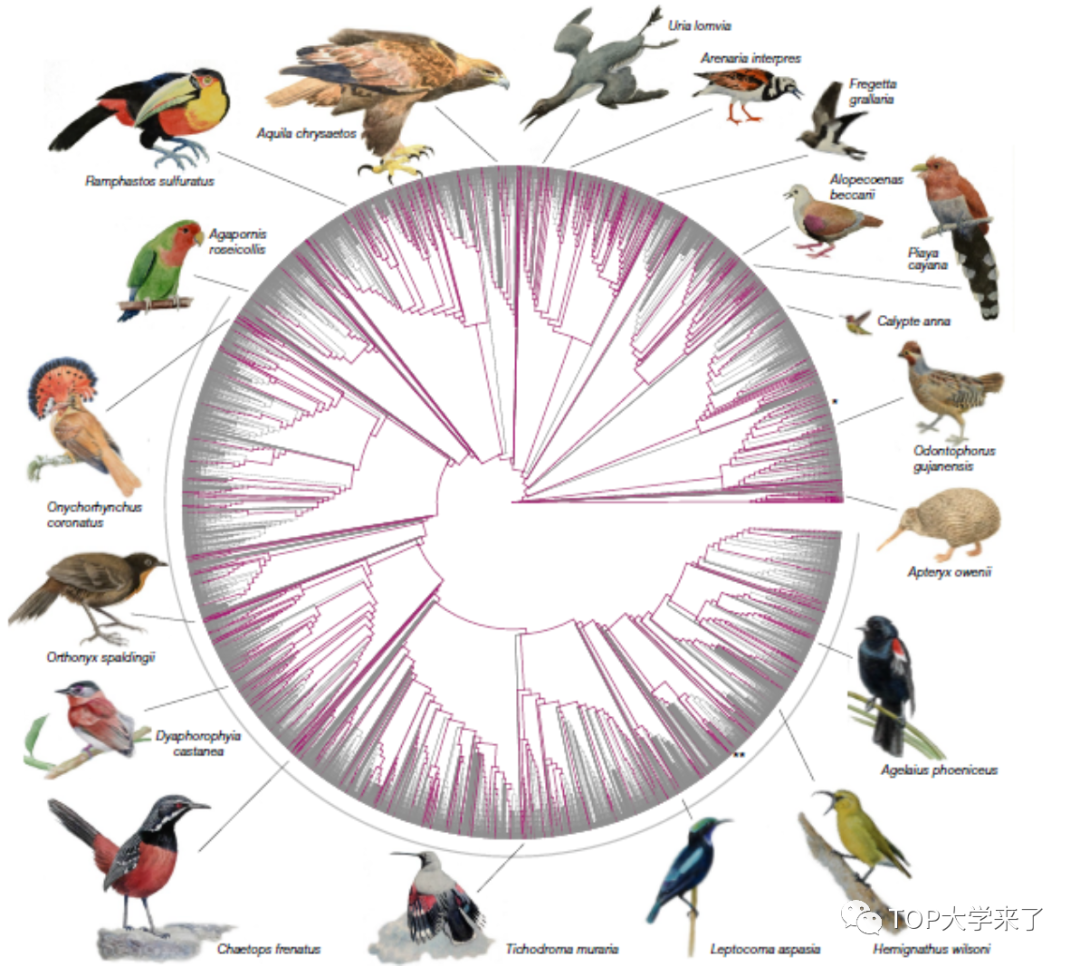
在第二篇论文中,研究通过Cactus算法分析了363个鸟类基因组数据,建立了更完善的同源基因集合。在比较研究中使用的基因组多样性的增加可以揭示更多的共享和谱系特异性变异,并改善对基因组特征的研究。这一基因组资源为跨物种比较分析中提供了新的视角,并有助于保护物种。




